

Predicting the impact of non-coding variants on DNA methylation
- PMID: 28334830
- PMCID: PMC5499808
- DOI: 10.1093/nar/gkx177
Predicting the impact of non-coding variants on DNA methylation
Abstract
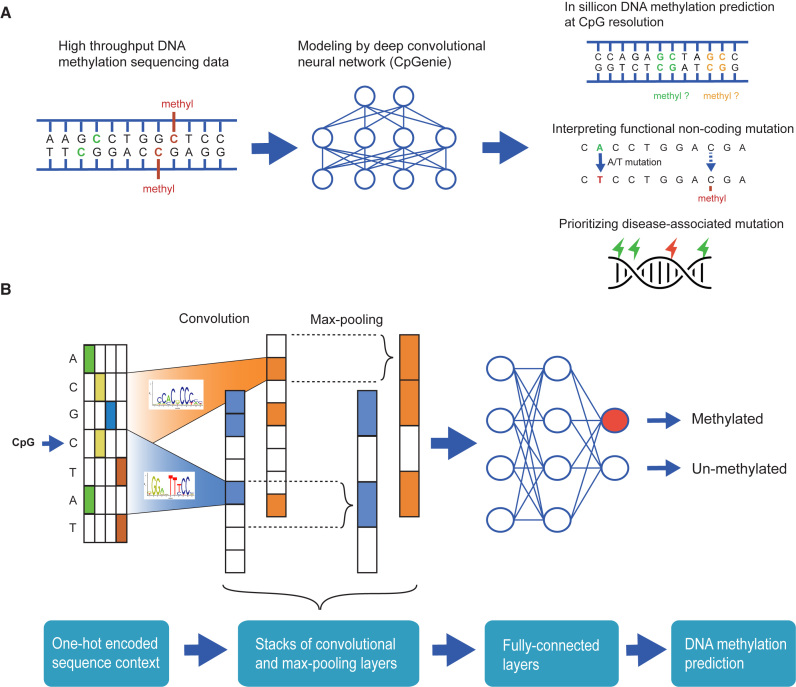
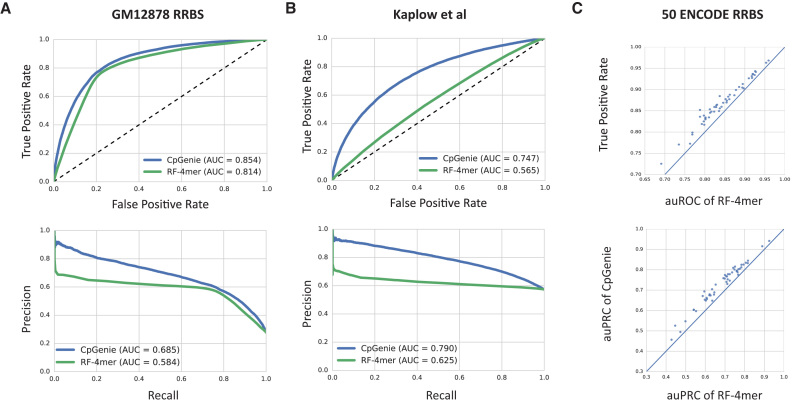
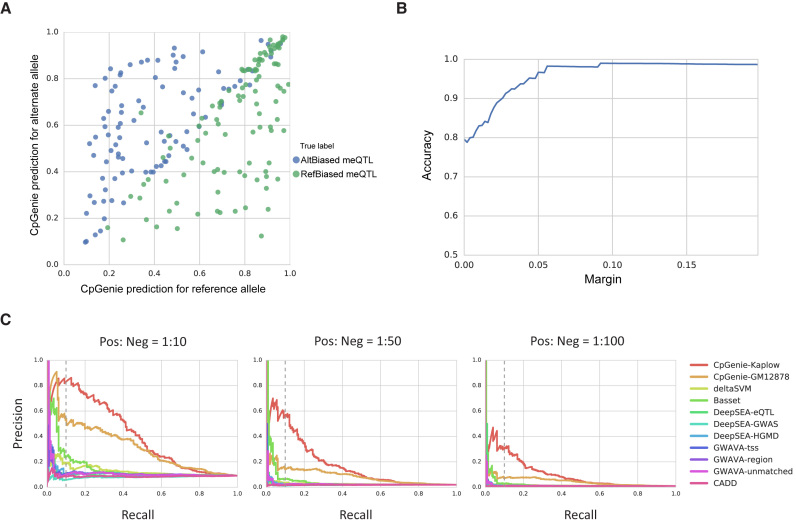
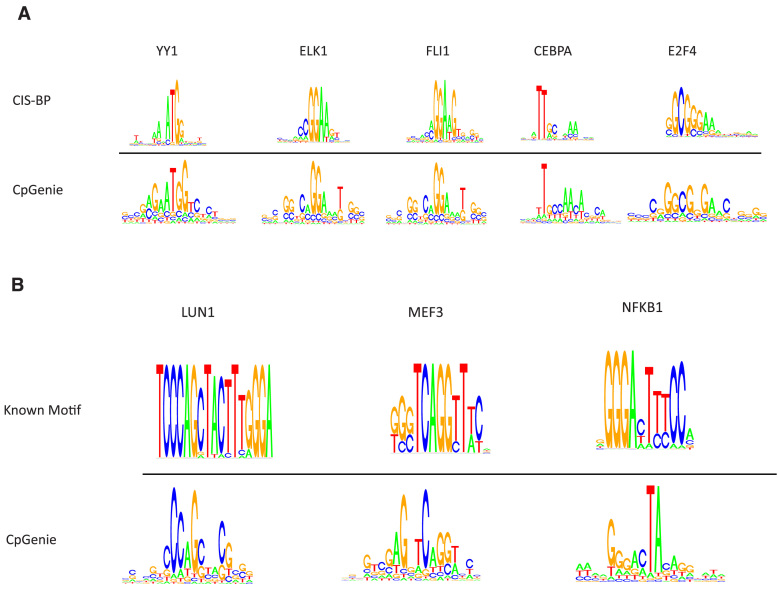
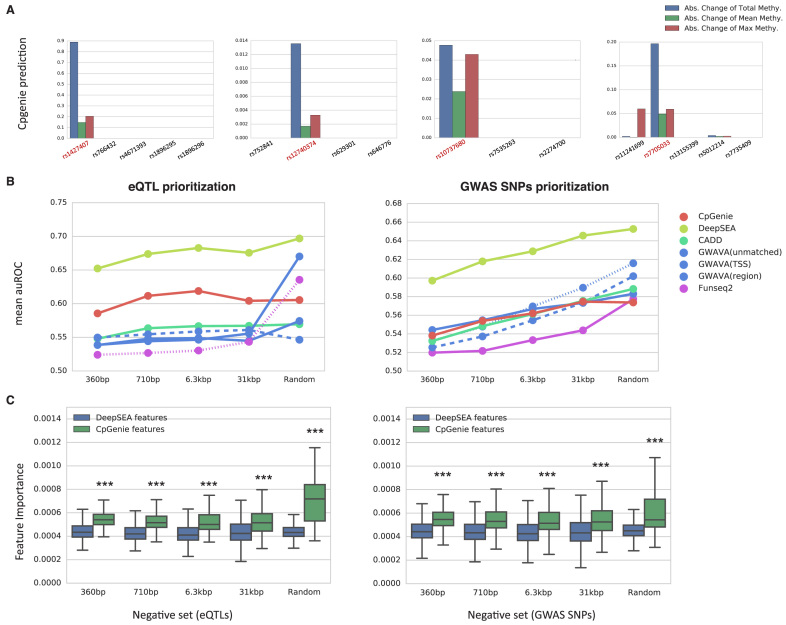
DNA methylation plays a crucial role in the establishment of tissue-specific gene expression and the regulation of key biological processes. However, our present inability to predict the effect of genome sequence variation on DNA methylation precludes a comprehensive assessment of the consequences of non-coding variation. We introduce CpGenie, a sequence-based framework that learns a regulatory code of DNA methylation using a deep convolutional neural network and uses this network to predict the impact of sequence variation on proximal CpG site DNA methylation. CpGenie produces allele-specific DNA methylation prediction with single-nucleotide sensitivity that enables accurate prediction of methylation quantitative trait loci (meQTL). We demonstrate that CpGenie prioritizes validated GWAS SNPs, and contributes to the prediction of functional non-coding variants, including expression quantitative trait loci (eQTL) and disease-associated mutations. CpGenie is publicly available to assist in identifying and interpreting regulatory non-coding variants.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
