

Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome
- PMID: 28334972
- PMCID: PMC5499761
- DOI: 10.1093/nar/gkx143
Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome
Abstract
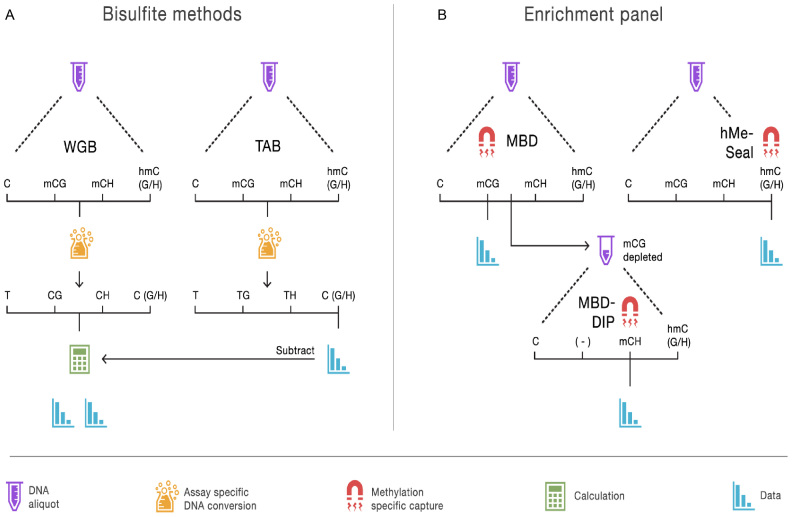
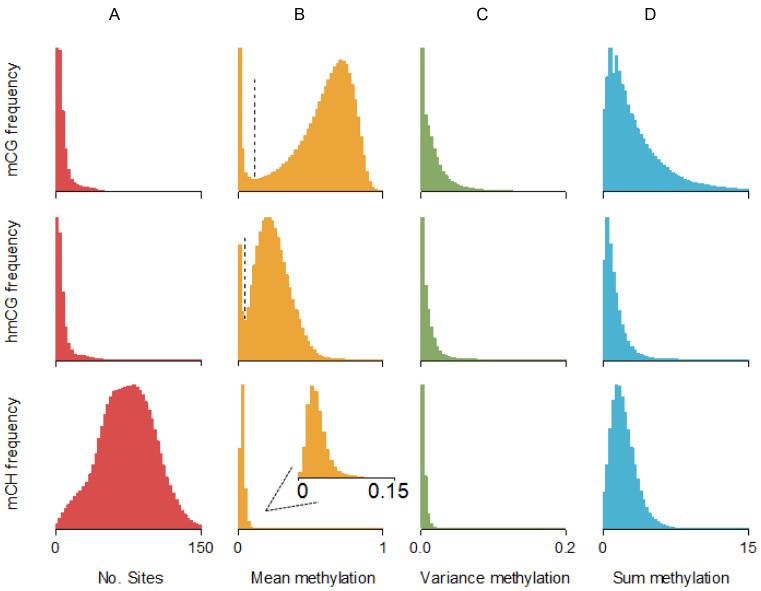
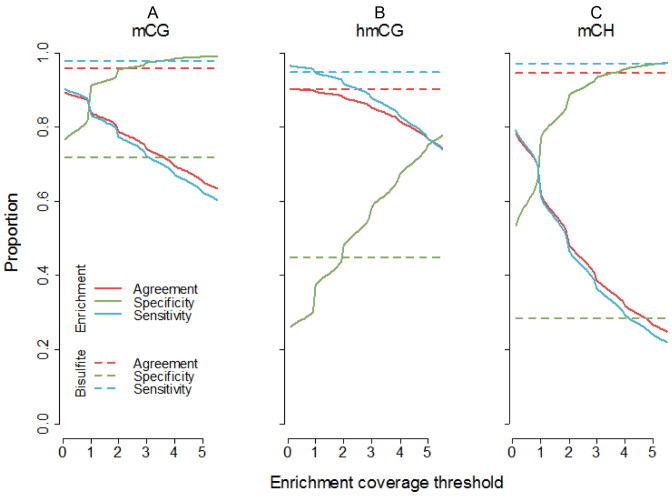
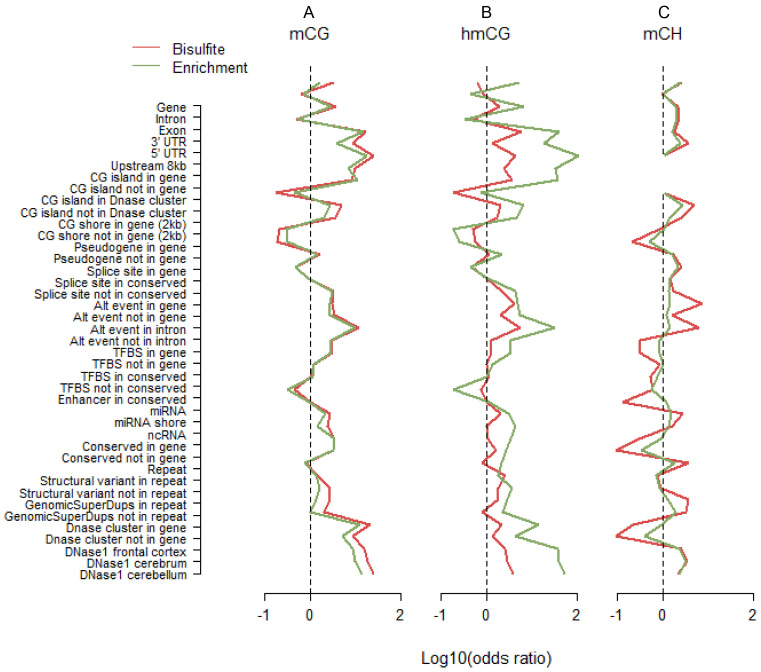
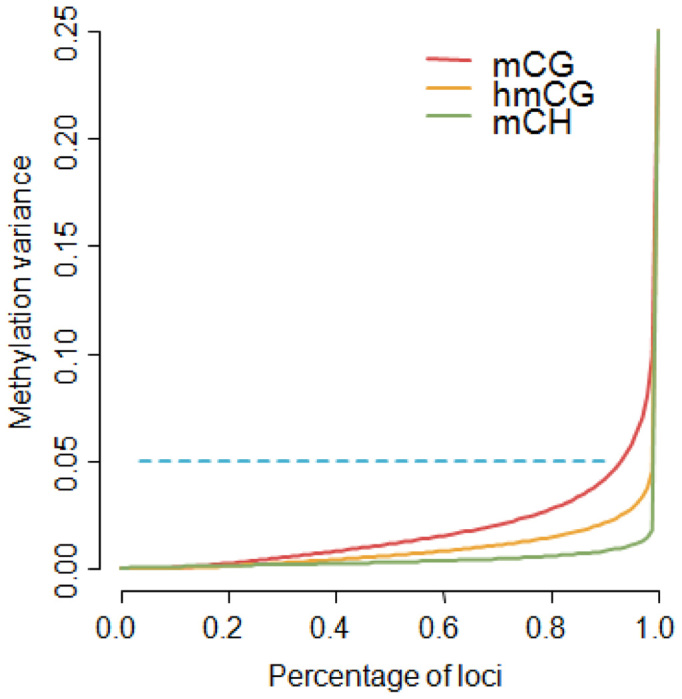
Methylome-wide association studies are typically performed using microarray technologies that only assay a very small fraction of the CG methylome and entirely miss two forms of methylation that are common in brain and likely of particular relevance for neuroscience and psychiatric disorders. The alternative is to use whole genome bisulfite (WGB) sequencing but this approach is not yet practically feasible with sample sizes required for adequate statistical power. We argue for revisiting methylation enrichment methods that, provided optimal protocols are used, enable comprehensive, adequately powered and cost-effective genome-wide investigations of the brain methylome. To support our claim we use data showing that enrichment methods approximate the sensitivity obtained with WGB methods and with slightly better specificity. However, this performance is achieved at <5% of the reagent costs. Furthermore, because many more samples can be sequenced simultaneously, projects can be completed about 15 times faster. Currently the only viable option available for comprehensive brain methylome studies, enrichment methods may be critical for moving the field forward.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Gage F.H., Temple S.. Neural stem cells: generating and regenerating the brain. Neuron. 2013; 80:588–601. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
