

Defining the 5΄ and 3΄ landscape of the Drosophila transcriptome with Exo-seq and RNaseH-seq
- PMID: 28335028
- PMCID: PMC5499799
- DOI: 10.1093/nar/gkx133
Defining the 5΄ and 3΄ landscape of the Drosophila transcriptome with Exo-seq and RNaseH-seq
Abstract
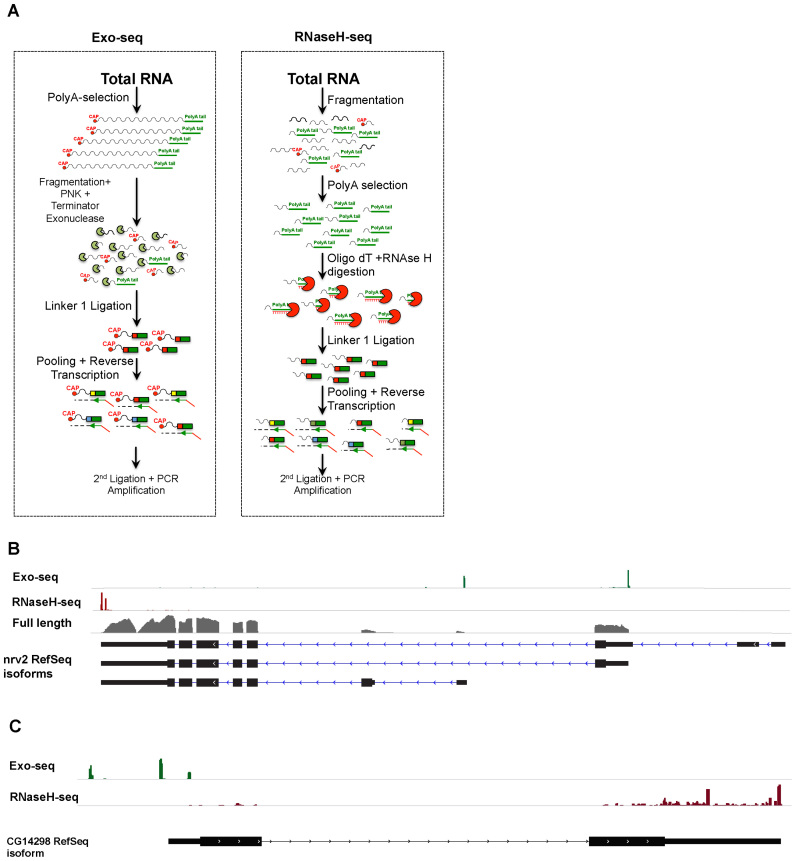
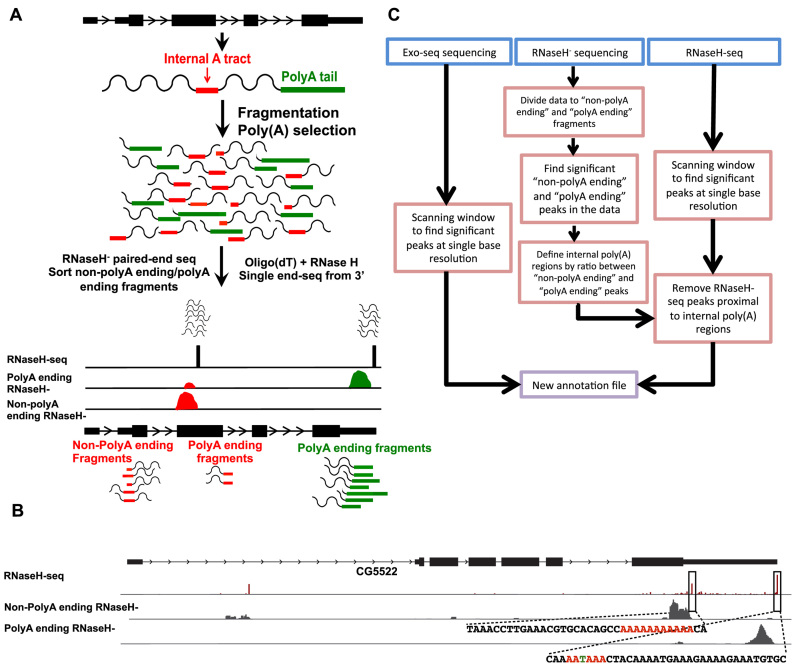
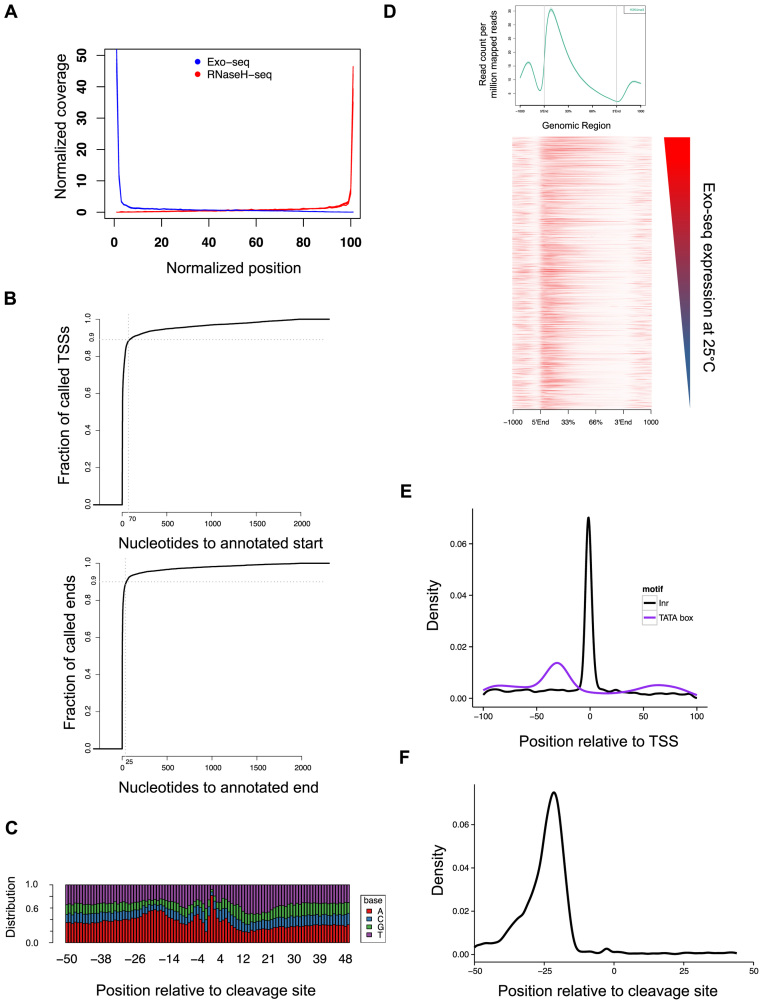
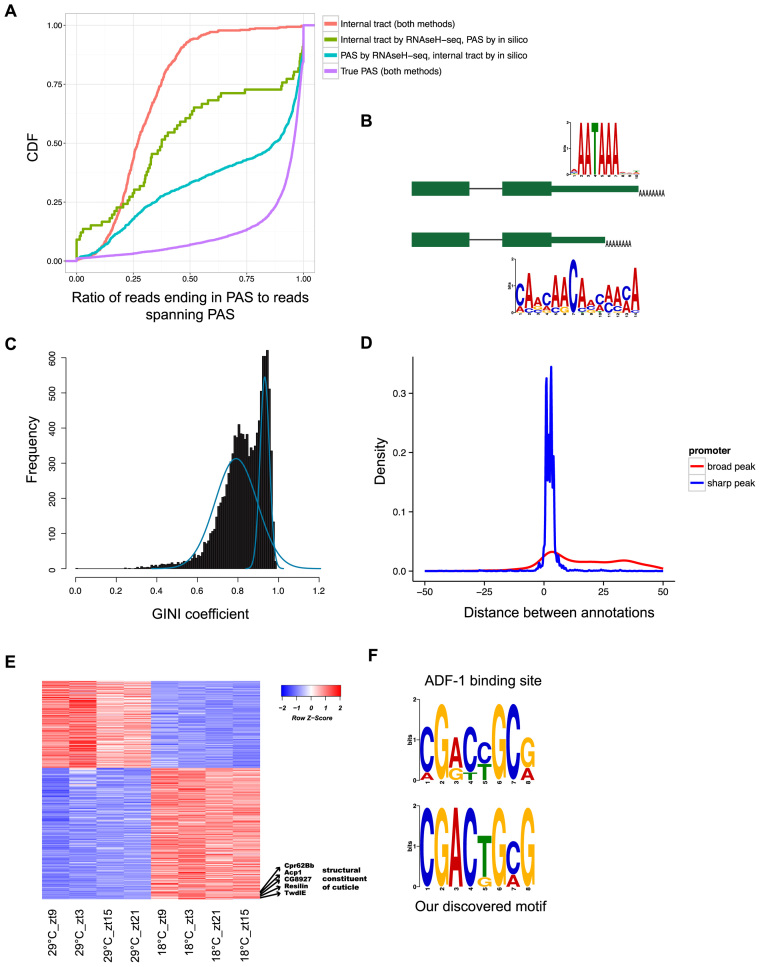
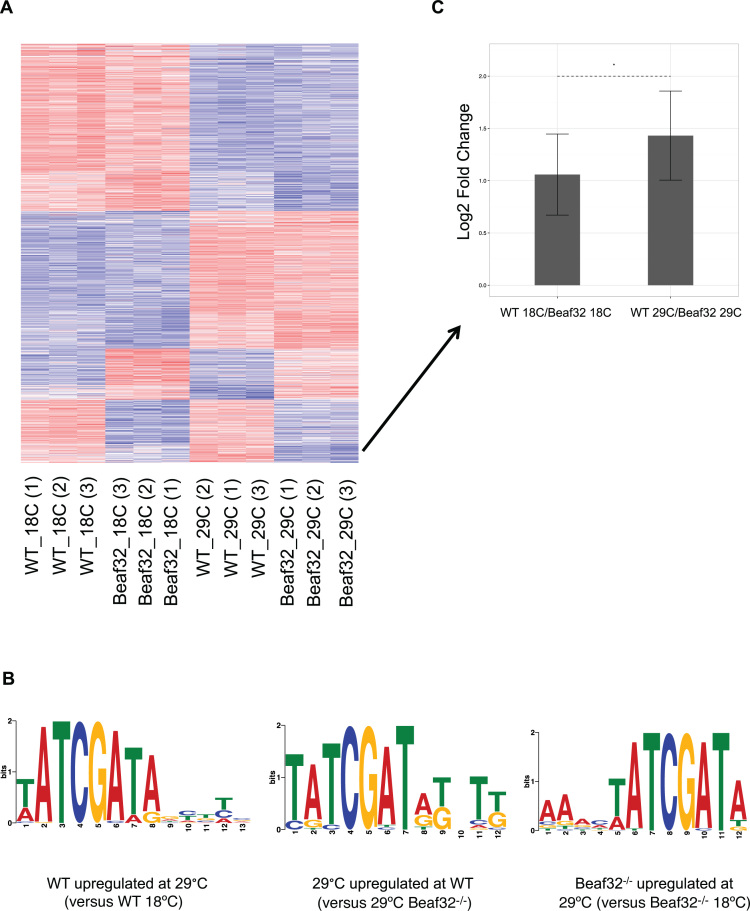
Cells regulate biological responses in part through changes in transcription start sites (TSS) or cleavage and polyadenylation sites (PAS). To fully understand gene regulatory networks, it is therefore critical to accurately annotate cell type-specific TSS and PAS. Here we present a simple and straightforward approach for genome-wide annotation of 5΄- and 3΄-RNA ends. Our approach reliably discerns bona fide PAS from false PAS that arise due to internal poly(A) tracts, a common problem with current PAS annotation methods. We applied our methodology to study the impact of temperature on the Drosophila melanogaster head transcriptome. We found hundreds of previously unidentified TSS and PAS which revealed two interesting phenomena: first, genes with multiple PASs tend to harbor a motif near the most proximal PAS, which likely represents a new cleavage and polyadenylation signal. Second, motif analysis of promoters of genes affected by temperature suggested that boundary element association factor of 32 kDa (BEAF-32) and DREF mediates a transcriptional program at warm temperatures, a result we validated in a fly line where beaf-32 is downregulated. These results demonstrate the utility of a high-throughput platform for complete experimental and computational analysis of mRNA-ends to improve gene annotation.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Moore M.J. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005; 309:1514–1518. - PubMed
-
- Carninci P., Kasukawa T., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C.. The transcriptional landscape of the mammalian genome. Science. 2005; 309:1559–1563. - PubMed
-
- Davuluri R. V, Suzuki Y., Sugano S., Plass C., Huang T.H.-M.. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008; 24:167–177. - PubMed
-
- Elkon R., Ugalde A.P., Agami R.. Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 2013; 14:496–506. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
