

A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions
- PMID: 28335035
- PMCID: PMC5411739
- DOI: 10.1093/hmg/ddx060
A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions
Erratum in
-
A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions.Hum Mol Genet. 2017 May 1;26(9):1786. doi: 10.1093/hmg/ddx130. Hum Mol Genet. 2017. PMID: 28475771 Free PMC article. No abstract available.
Abstract
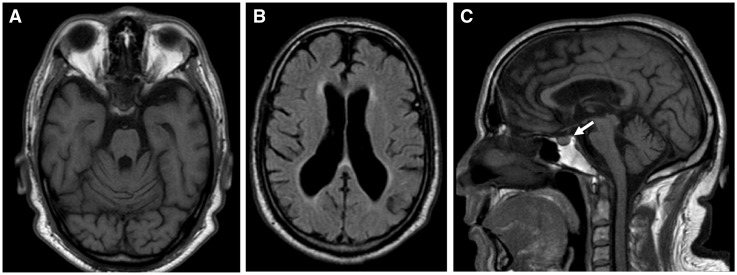
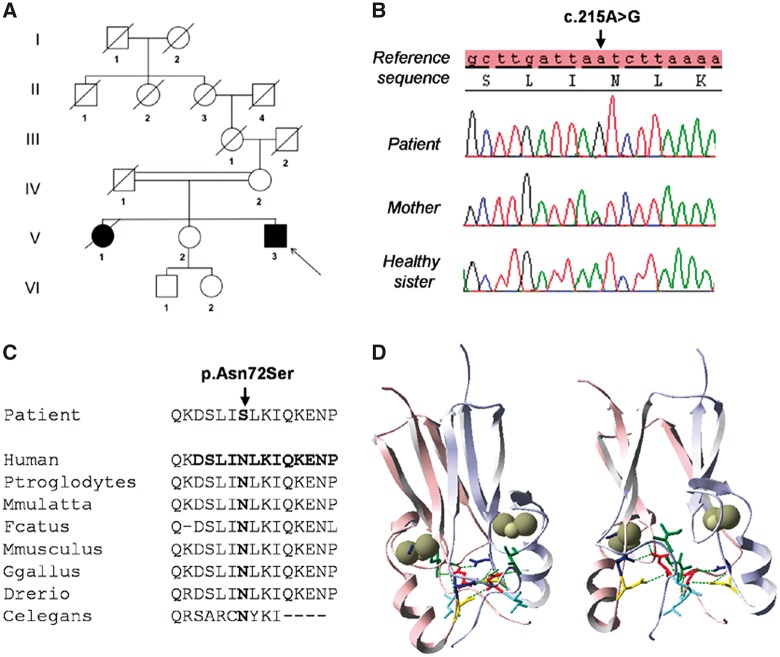
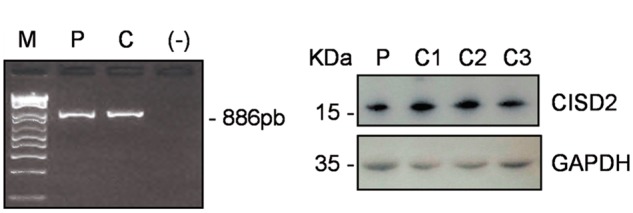
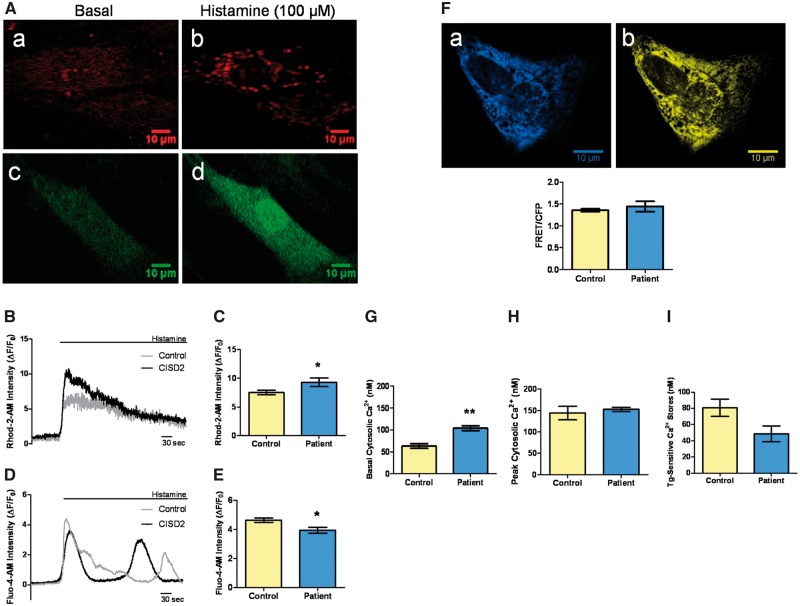
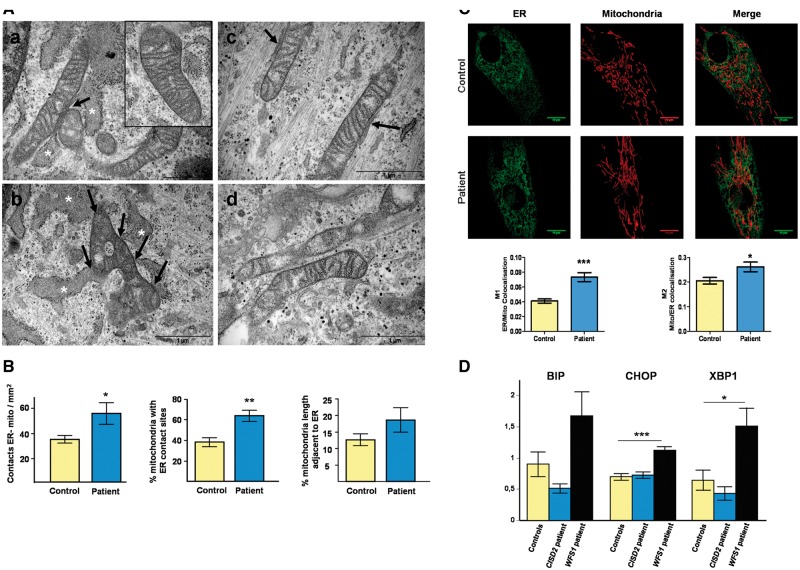
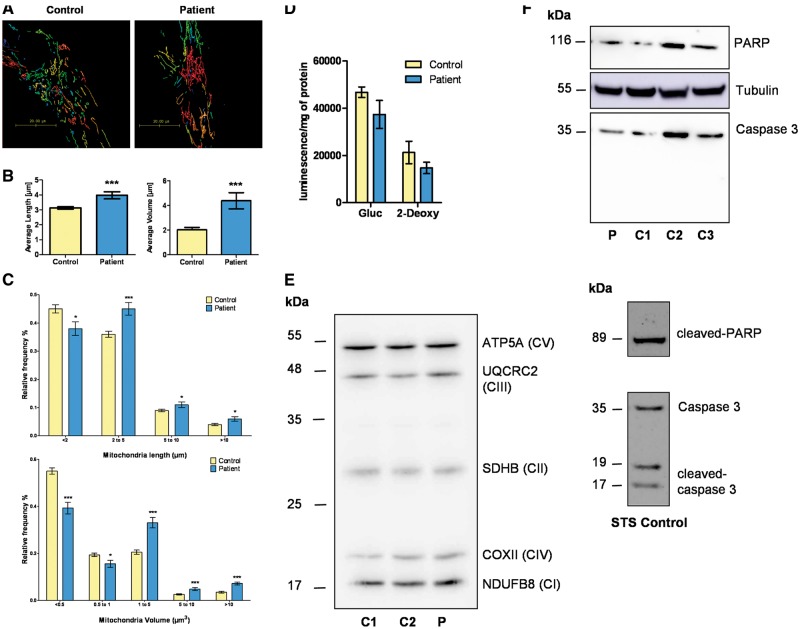
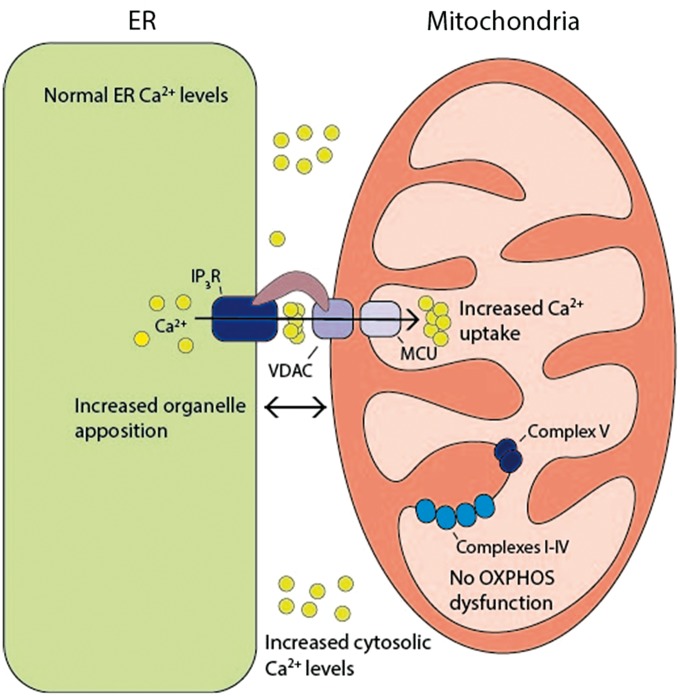
Wolfram syndrome (WS) is a progressive neurodegenerative disease characterized by early-onset optic atrophy and diabetes mellitus, which can be associated with more extensive central nervous system and endocrine complications. The majority of patients harbour pathogenic WFS1 mutations, but recessive mutations in a second gene, CISD2, have been described in a small number of families with Wolfram syndrome type 2 (WFS2). The defining diagnostic criteria for WFS2 also consist of optic atrophy and diabetes mellitus, but unlike WFS1, this phenotypic subgroup has been associated with peptic ulcer disease and an increased bleeding tendency. Here, we report on a novel homozygous CISD2 mutation (c.215A > G; p.Asn72Ser) in a Moroccan patient with an overlapping phenotype suggesting that Wolfram syndrome type 1 and type 2 form a continuous clinical spectrum with genetic heterogeneity. The present study provides strong evidence that this particular CISD2 mutation disturbs cellular Ca2+ homeostasis with enhanced Ca2+ flux from the ER to mitochondria and cytosolic Ca2+ abnormalities in patient-derived fibroblasts. This Ca2+ dysregulation was associated with increased ER-mitochondria contact, a swollen ER lumen and a hyperfused mitochondrial network in the absence of overt ER stress. Although there was no marked alteration in mitochondrial bioenergetics under basal conditions, culture of patient-derived fibroblasts in glucose-free galactose medium revealed a respiratory chain defect in complexes I and II, and a trend towards decreased ATP levels. Our results provide important novel insight into the potential disease mechanisms underlying the neurodegenerative consequences of CISD2 mutations and the subsequent development of multisystemic disease.
© The Author 2017. Published by Oxford University Press.
Figures
References
-
- Barrett T.G., Bundey S.E., Macleod A.F. (1995) Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet Lond. Engl., 346, 1458–1463. - PubMed
-
- Khanim F., Kirk J., Latif F., Barrett T.G. (2001) WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum. Mutat., 17, 357–367. - PubMed
-
- Rondinelli M., Novara F., Calcaterra V., Zuffardi O., Genovese S. (2015) Wolfram syndrome 2: a novel CISD2 mutation identified in Italian siblings. Acta Diabetol., 52, 175–178. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
