

NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury
- PMID: 28340575
- PMCID: PMC5366128
- DOI: 10.1186/s12974-017-0843-4
NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury
Abstract
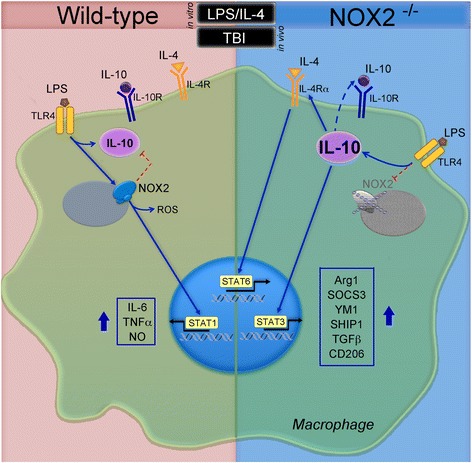
Background: NADPH oxidase (NOX2) is an enzyme system that generates reactive oxygen species (ROS) in microglia and macrophages. Excessive ROS production is linked with neuroinflammation and chronic neurodegeneration following traumatic brain injury (TBI). Redox signaling regulates macrophage/microglial phenotypic responses (pro-inflammatory versus anti-inflammatory), and NOX2 inhibition following moderate-to-severe TBI markedly reduces pro-inflammatory activation of macrophages/microglia resulting in concomitant increases in anti-inflammatory responses. Here, we report the signaling pathways that regulate NOX2-dependent macrophage/microglial phenotype switching in the TBI brain.
Methods: Bone marrow-derived macrophages (BMDMs) prepared from wildtype (C57Bl/6) and NOX2 deficient (NOX2-/-) mice were treated with lipopolysaccharide (LPS; 10 ng/ml), interleukin-4 (IL-4; 10 ng/ml), or combined LPS/IL-4 to investigate signal transduction pathways associated with macrophage activation using western immunoblotting and qPCR analyses. Signaling pathways and activation markers were evaluated in ipsilateral cortical tissue obtained from adult male wildtype and NOX2-/- mice that received moderate-level controlled cortical impact (CCI). A neutralizing anti-IL-10 approach was used to determine the effects of IL-10 on NOX2-dependent transitions from pro- to anti-inflammatory activation states.
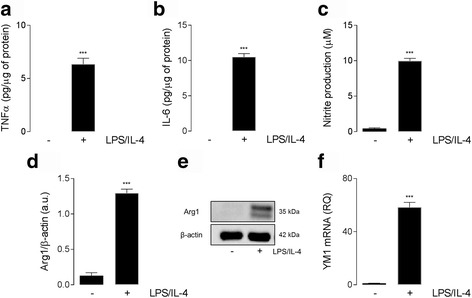
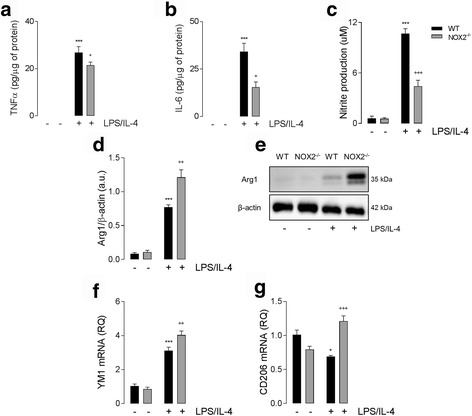
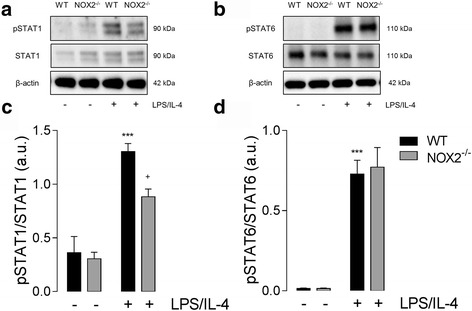
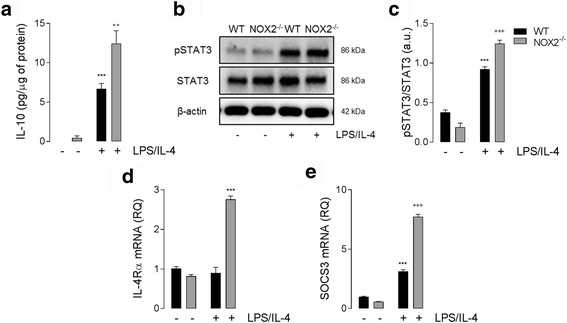
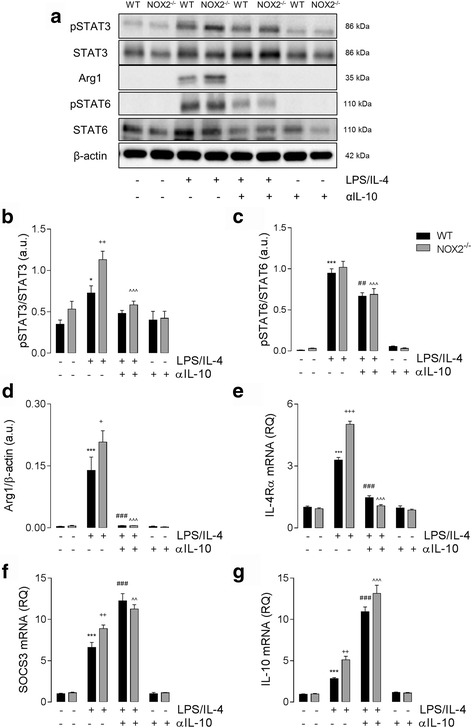
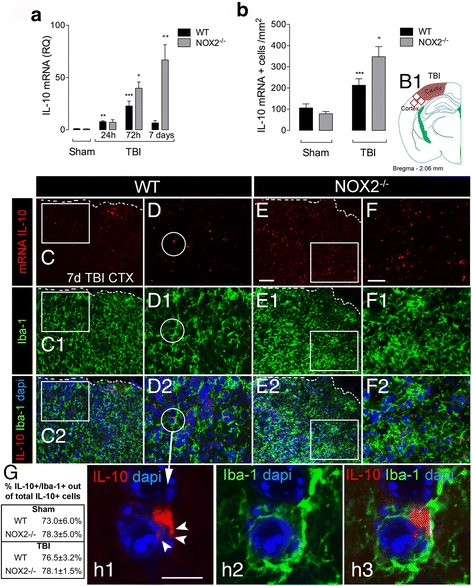
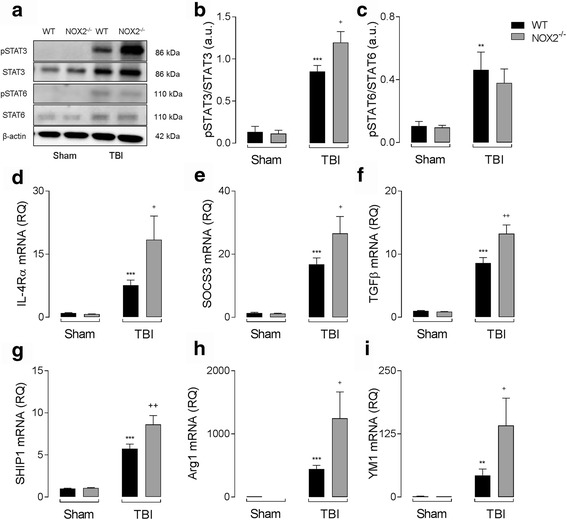
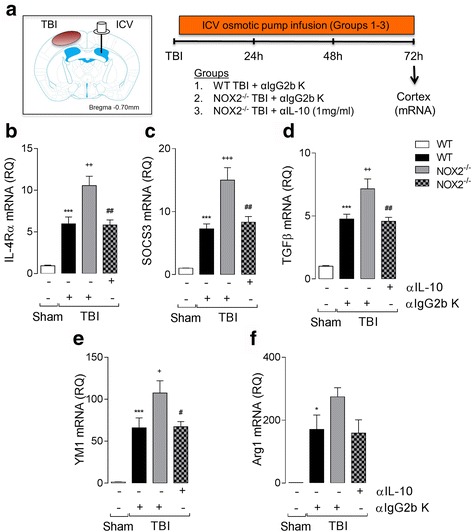
Results: Using an LPS/IL-4-stimulated BMDM model that mimics the mixed pro- and anti-inflammatory responses observed in the injured cortex, we show that NOX2-/- significantly reduces STAT1 signaling and markers of pro-inflammatory activation. In addition, NOX2-/- BMDMs significantly increase anti-inflammatory marker expression; IL-10-mediated STAT3 signaling, but not STAT6 signaling, appears to be critical in regulating this anti-inflammatory response. Following moderate-level CCI, IL-10 is significantly increased in microglia/macrophages in the injured cortex of NOX2-/- mice. These changes are associated with increased STAT3 activation, but not STAT6 activation, and a robust anti-inflammatory response. Neutralization of IL-10 in NOX2-/- BMDMs or CCI mice blocks STAT3 activation and the anti-inflammatory response, thereby demonstrating a critical role for IL-10 in regulating NOX2-dependent transitions between pro- and anti-inflammatory activation states.
Conclusions: These studies indicate that following TBI NOX2 inhibition promotes a robust anti-inflammatory response in macrophages/microglia that is mediated by the IL-10/STAT3 signaling pathway. Thus, therapeutic interventions that inhibit macrophage/microglial NOX2 activity may improve TBI outcomes by not only limiting pro-inflammatory neurotoxic responses, but also enhancing IL-10-mediated anti-inflammatory responses that are neuroprotective.
Keywords: Interleukin-10; Macrophage; NADPH oxidase; NOX2; Neuroinflammation; Traumatic brain injury.
Figures
References
-
- Ohmori Y, Hamilton TA. Requirement for STAT1 in LPS-induced gene expression in macrophages. J Leukoc Biol. 2001;69:598–604. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
