

Metabolic Dysfunction in Parkinson's Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism
- PMID: 28341600
- PMCID: PMC5555796
- DOI: 10.1016/j.brainresbull.2017.03.009
Metabolic Dysfunction in Parkinson's Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism
Abstract
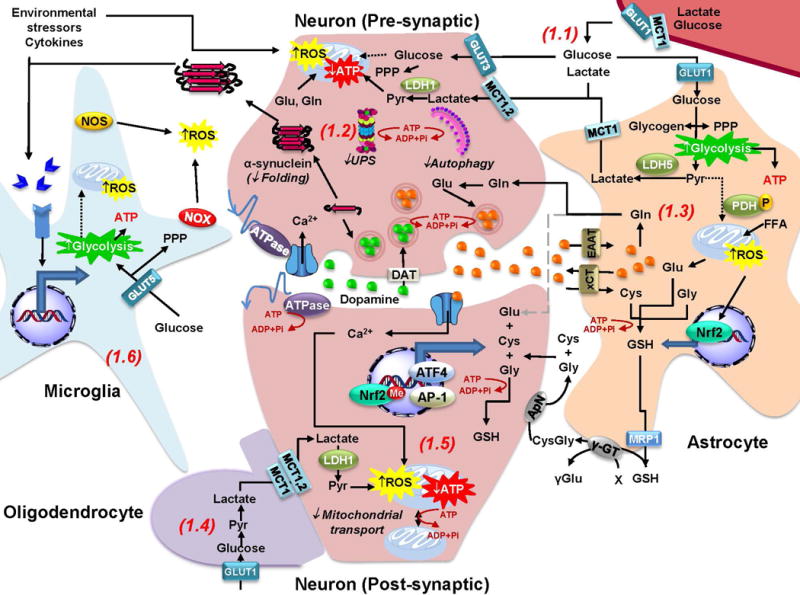
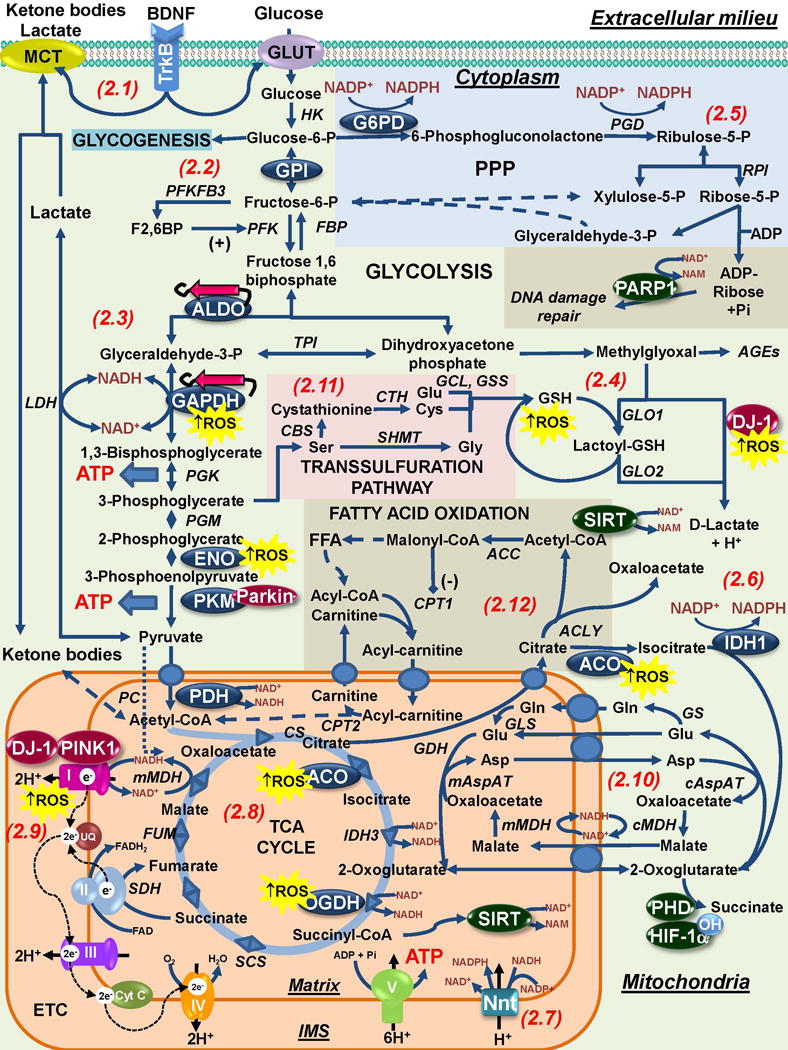
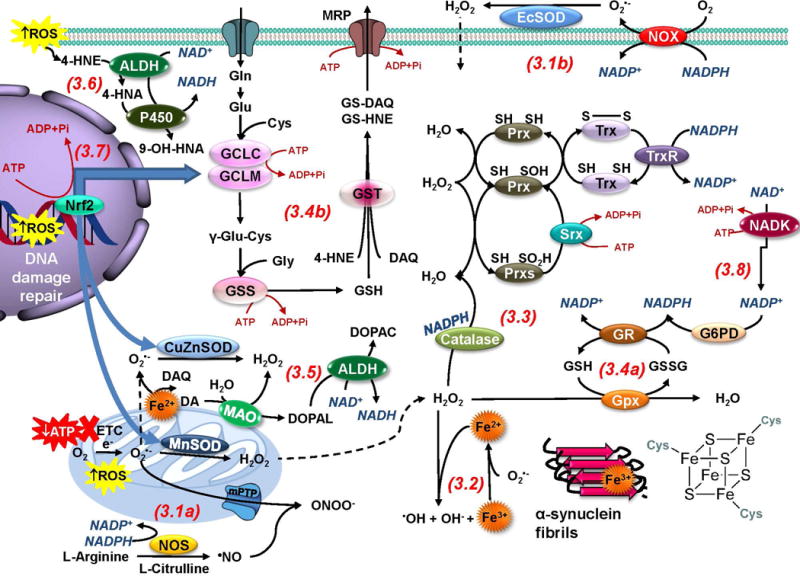
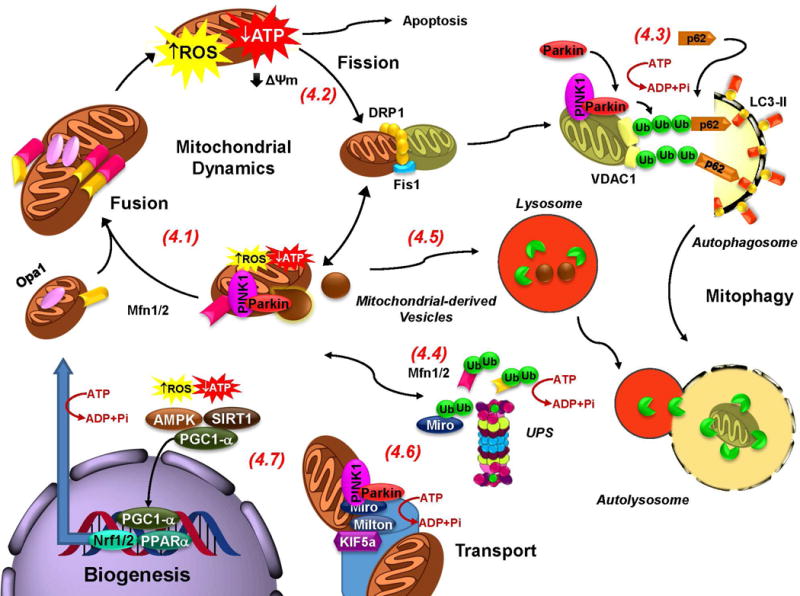
The loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the accumulation of protein inclusions (Lewy bodies) are the pathological hallmarks of Parkinson's disease (PD). PD is triggered by genetic alterations, environmental/occupational exposures and aging. However, the exact molecular mechanisms linking these PD risk factors to neuronal dysfunction are still unclear. Alterations in redox homeostasis and bioenergetics (energy failure) are thought to be central components of neurodegeneration that contribute to the impairment of important homeostatic processes in dopaminergic cells such as protein quality control mechanisms, neurotransmitter release/metabolism, axonal transport of vesicles and cell survival. Importantly, both bioenergetics and redox homeostasis are coupled to neuro-glial central carbon metabolism. We and others have recently established a link between the alterations in central carbon metabolism induced by PD risk factors, redox homeostasis and bioenergetics and their contribution to the survival/death of dopaminergic cells. In this review, we focus on the link between metabolic dysfunction, energy failure and redox imbalance in PD, making an emphasis in the contribution of central carbon (glucose) metabolism. The evidence summarized here strongly supports the consideration of PD as a disorder of cell metabolism.
Keywords: Bioenergetics; Glucose; Glycolysis; Mitochondria; Neurodegeneration; Oxidative stress; TCA cycle.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med. 1993;328:176–183. - PubMed
-
- Abdin AA, Hamouda HE. Mechanism of the neuroprotective role of coenzyme Q10 with or without L-dopa in rotenone-induced parkinsonism. Neuropharmacology. 2008;55:1340–1346. - PubMed
-
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006;7:207–219. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
