

Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics
- PMID: 28341641
- PMCID: PMC5379987
- DOI: 10.1084/jem.20170193
Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics
Abstract
A latent reservoir for HIV-1 in resting CD4+ T lymphocytes precludes cure. Mechanisms underlying reservoir stability are unclear. Recent studies suggest an unexpected degree of infected cell proliferation in vivo. T cell activation drives proliferation but also reverses latency, resulting in productive infection that generally leads to cell death. In this study, we show that latently infected cells can proliferate in response to mitogens without producing virus, generating progeny cells that can release infectious virus. Thus, assays relying on one round of activation underestimate reservoir size. Sequencing of independent clonal isolates of replication-competent virus revealed that 57% had env sequences identical to other isolates from the same patient. Identity was confirmed by full-genome sequencing and was not attributable to limited viral diversity. Phylogenetic and statistical analysis suggested that identical sequences arose from in vivo proliferation of infected cells, rather than infection of multiple cells by a dominant viral species. The possibility that much of the reservoir arises by cell proliferation presents challenges to cure.
© 2017 Hosmane et al.
Figures
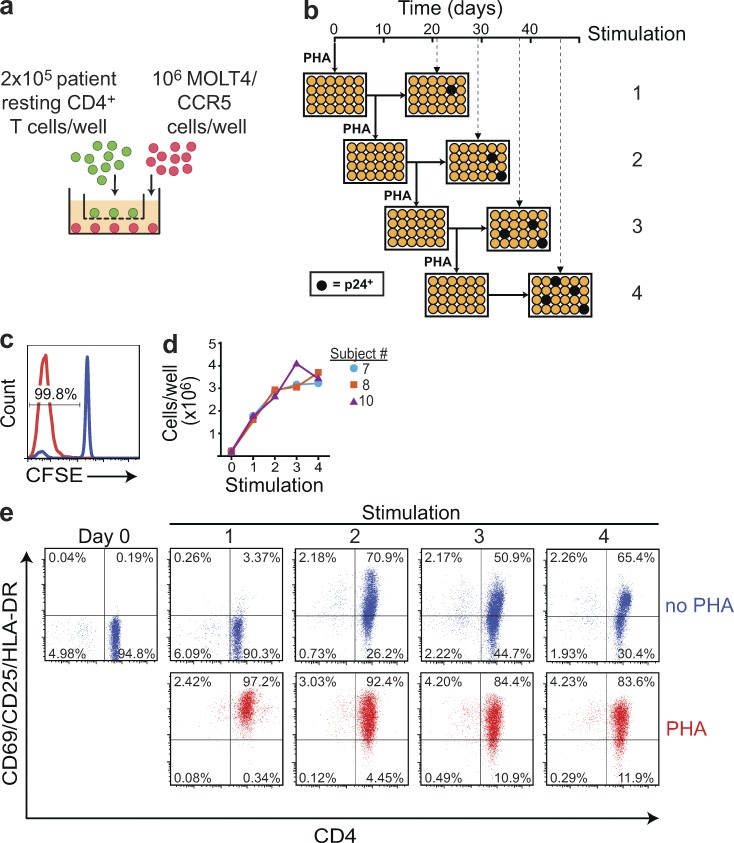
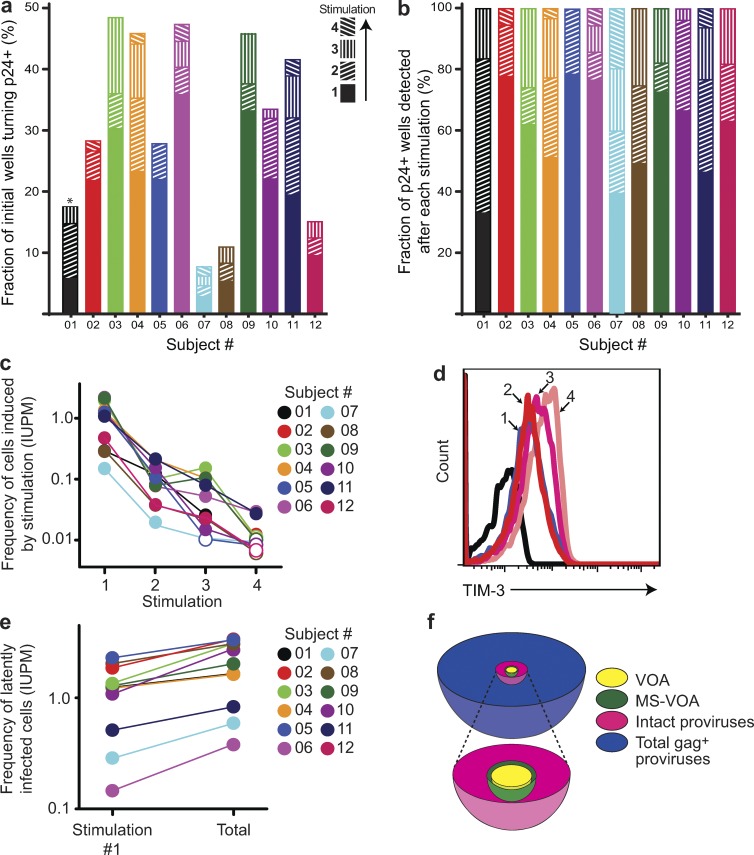
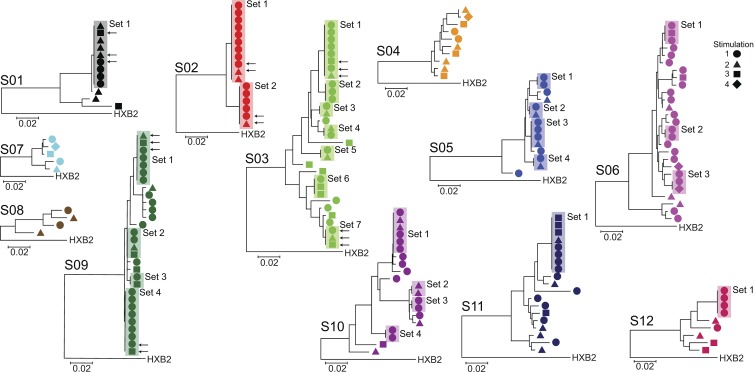
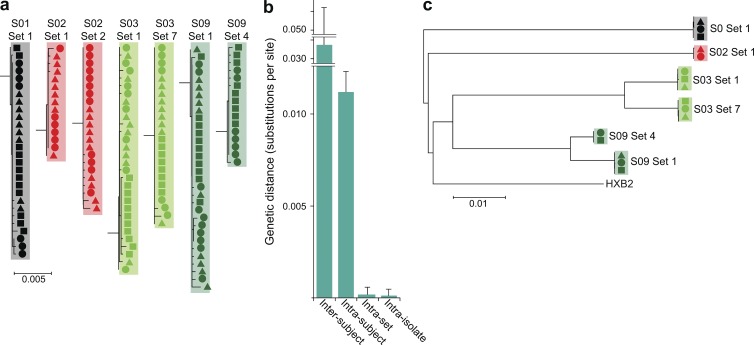
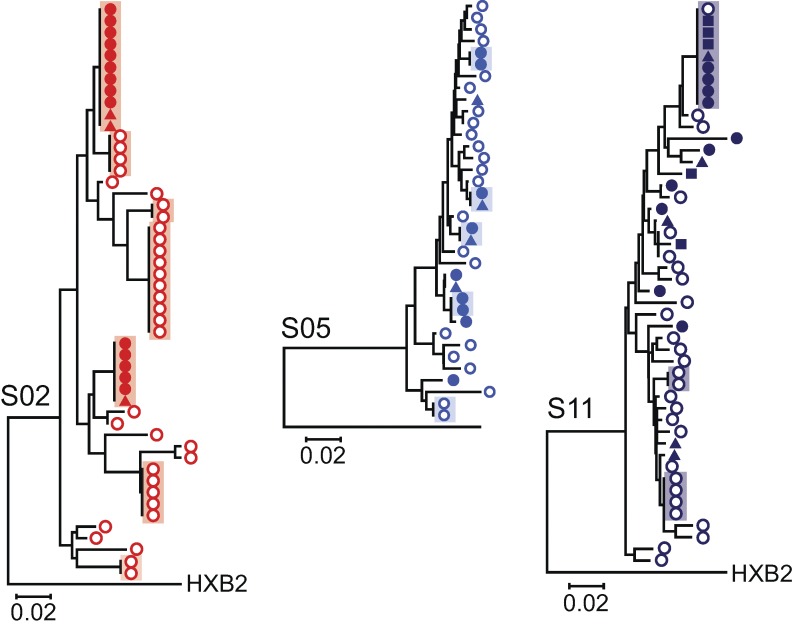
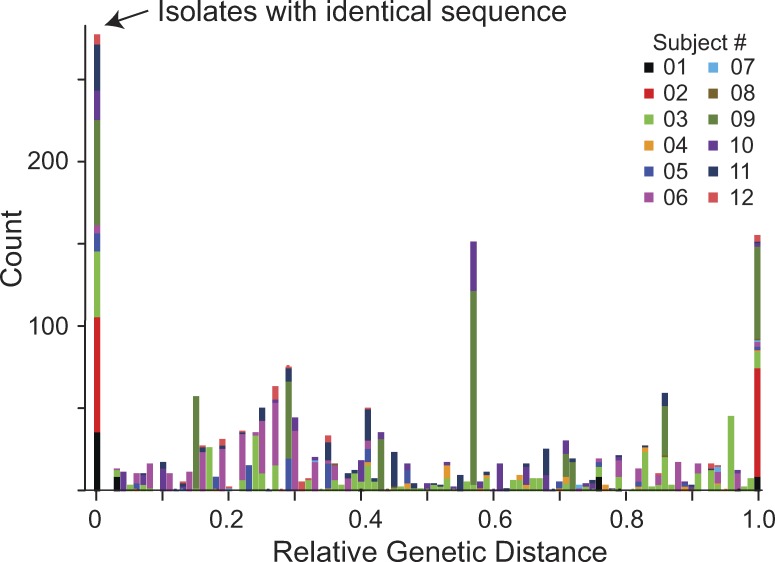
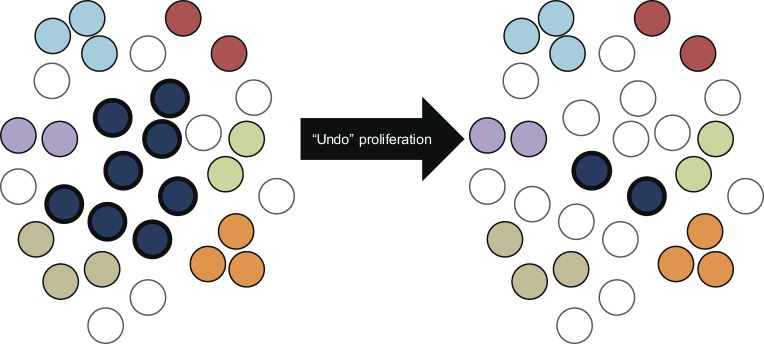
Comment in
-
HIV: Persistence through division.J Exp Med. 2017 Apr 3;214(4):875-876. doi: 10.1084/jem.20170463. Epub 2017 Mar 27. J Exp Med. 2017. PMID: 28348204 Free PMC article.
References
-
- Archin N.M., Liberty A.L., Kashuba A.D., Choudhary S.K., Kuruc J.D., Crooks A.M., Parker D.C., Anderson E.M., Kearney M.F., Strain M.C., et al. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 487:482–485. 10.1038/nature11286 - DOI - PMC - PubMed
-
- Bailey J.R., Sedaghat A.R., Kieffer T., Brennan T., Lee P.K., Wind-Rotolo M., Haggerty C.M., Kamireddi A.R., Liu Y., Lee J., et al. 2006. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 80:6441–6457. 10.1128/JVI.00591-06 - DOI - PMC - PubMed
-
- Banga R., Procopio F.A., Noto A., Pollakis G., Cavassini M., Ohmiti K., Corpataux J.M., de Leval L., Pantaleo G., and Perreau M.. 2016. PD-1+ and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 22:754–761. 10.1038/nm.4113 - DOI - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
