

Mevalonate Cascade Inhibition by Simvastatin Induces the Intrinsic Apoptosis Pathway via Depletion of Isoprenoids in Tumor Cells
- PMID: 28344327
- PMCID: PMC5366866
- DOI: 10.1038/srep44841
Mevalonate Cascade Inhibition by Simvastatin Induces the Intrinsic Apoptosis Pathway via Depletion of Isoprenoids in Tumor Cells
Abstract
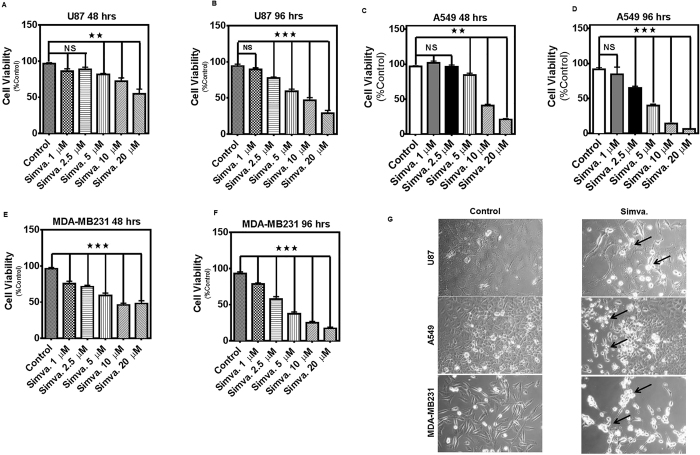
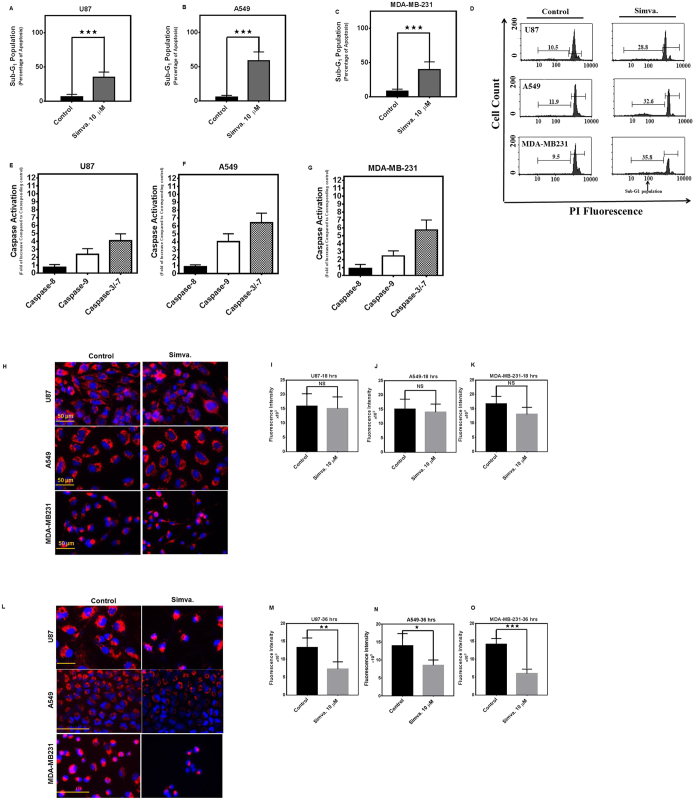
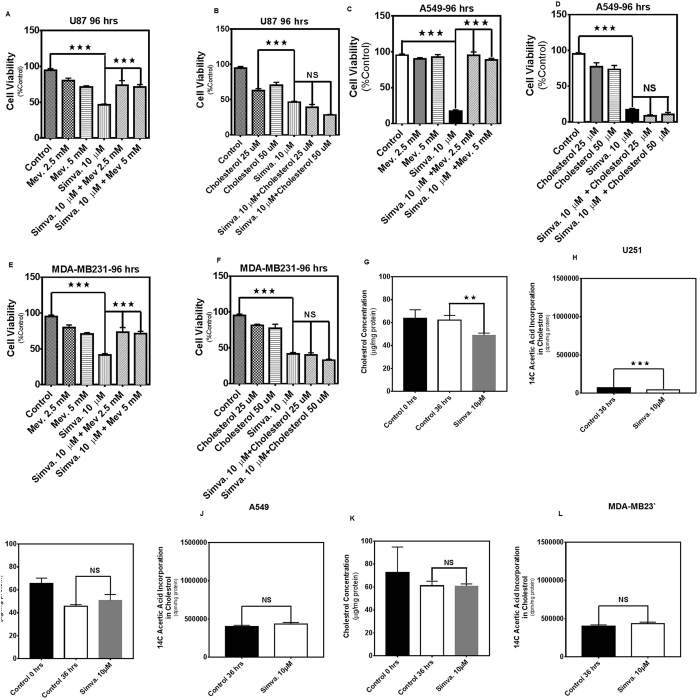
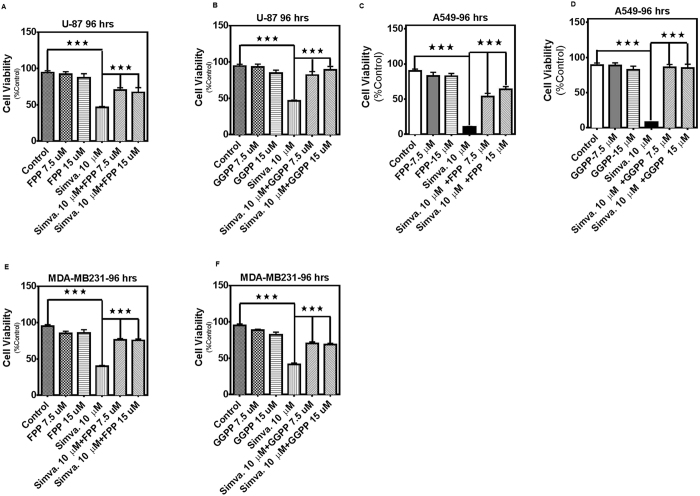
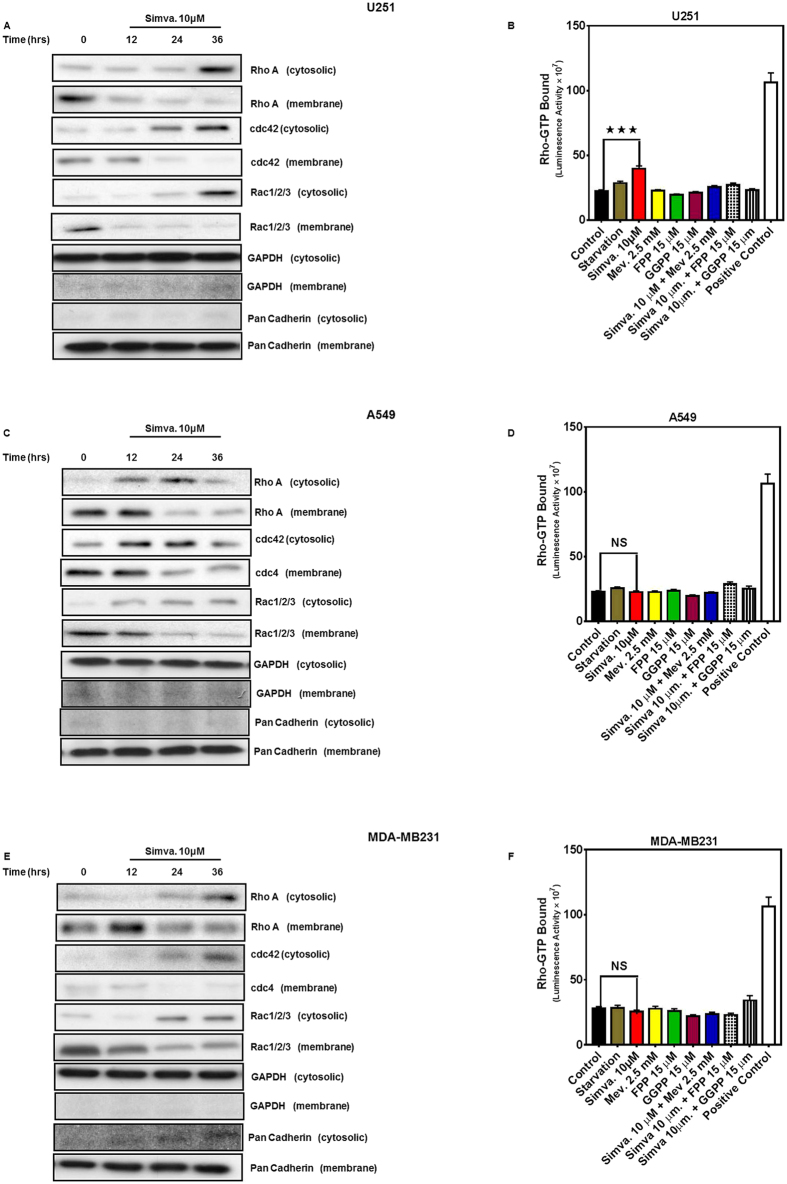
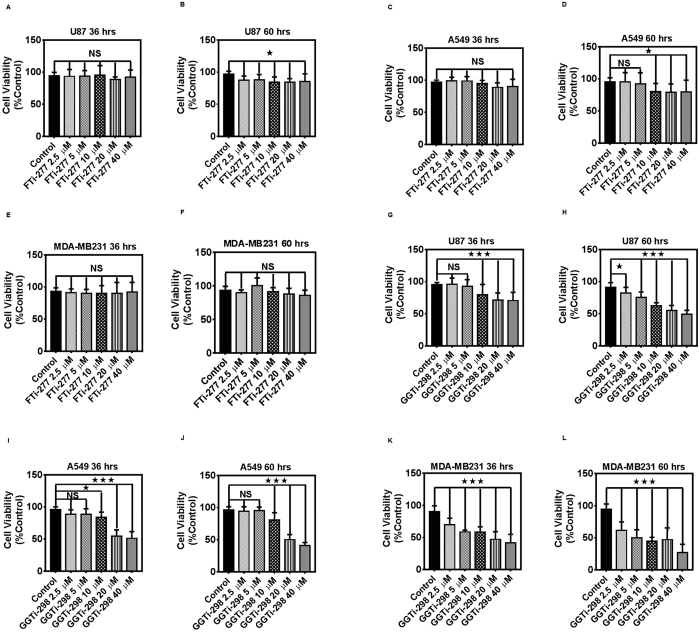
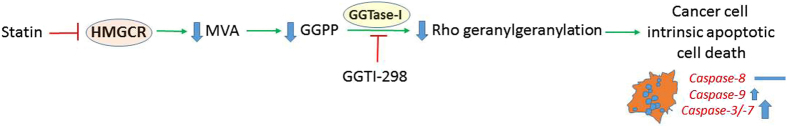
The mevalonate (MEV) cascade is responsible for cholesterol biosynthesis and the formation of the intermediate metabolites geranylgeranylpyrophosphate (GGPP) and farnesylpyrophosphate (FPP) used in the prenylation of proteins. Here we show that the MEV cascade inhibitor simvastatin induced significant cell death in a wide range of human tumor cell lines, including glioblastoma, astrocytoma, neuroblastoma, lung adenocarcinoma, and breast cancer. Simvastatin induced apoptotic cell death via the intrinsic apoptotic pathway. In all cancer cell types tested, simvastatin-induced cell death was not rescued by cholesterol, but was dependent on GGPP- and FPP-depletion. We confirmed that simvastatin caused the translocation of the small Rho GTPases RhoA, Cdc42, and Rac1/2/3 from cell membranes to the cytosol in U251 (glioblastoma), A549 (lung adenocarcinoma) and MDA-MB-231(breast cancer). Simvastatin-induced Rho-GTP loading significantly increased in U251 cells which were reversed with MEV, FPP, GGPP. In contrast, simvastatin did not change Rho-GTP loading in A549 and MDA-MB-231. Inhibition of geranylgeranyltransferase I by GGTi-298, but not farnesyltransferase by FTi-277, induced significant cell death in U251, A549, and MDA-MB-231. These results indicate that MEV cascade inhibition by simvastatin induced the intrinsic apoptosis pathway via inhibition of Rho family prenylation and depletion of GGPP, in a variety of different human cancer cell lines.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Zaidi N., Swinnen J. V. & Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer research 72, 3709–3714 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
