

Abnormal glycosylation in Joubert syndrome type 10
- PMID: 28344780
- PMCID: PMC5364566
- DOI: 10.1186/s13630-017-0048-6
Abnormal glycosylation in Joubert syndrome type 10
Abstract
Background: The discovery of disease pathogenesis requires systematic agnostic screening of multiple homeostatic processes that may become deregulated. We illustrate this principle in the evaluation and diagnosis of a 5-year-old boy with Joubert syndrome type 10 (JBTS10). He carried the OFD1 mutation p.Gln886Lysfs*2 (NM_003611.2: c.2656del) and manifested features of Joubert syndrome.
Methods: We integrated exome sequencing, MALDI-TOF mass spectrometry analyses of plasma and cultured dermal fibroblasts glycomes, and full clinical evaluation of the proband. Analyses of cilia formation and lectin staining were performed by immunofluorescence. Measurement of cellular nucleotide sugar levels was performed with high-performance anion-exchange chromatography with pulsed amperometric detection. Statistical analyses utilized the Student's and Fisher's exact t tests.
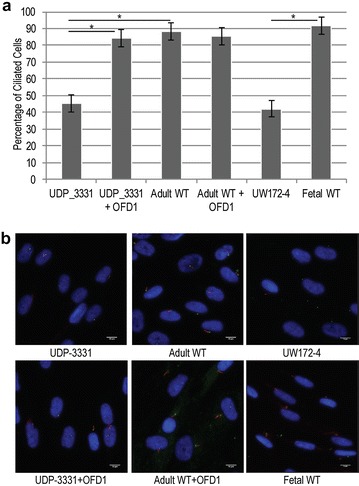
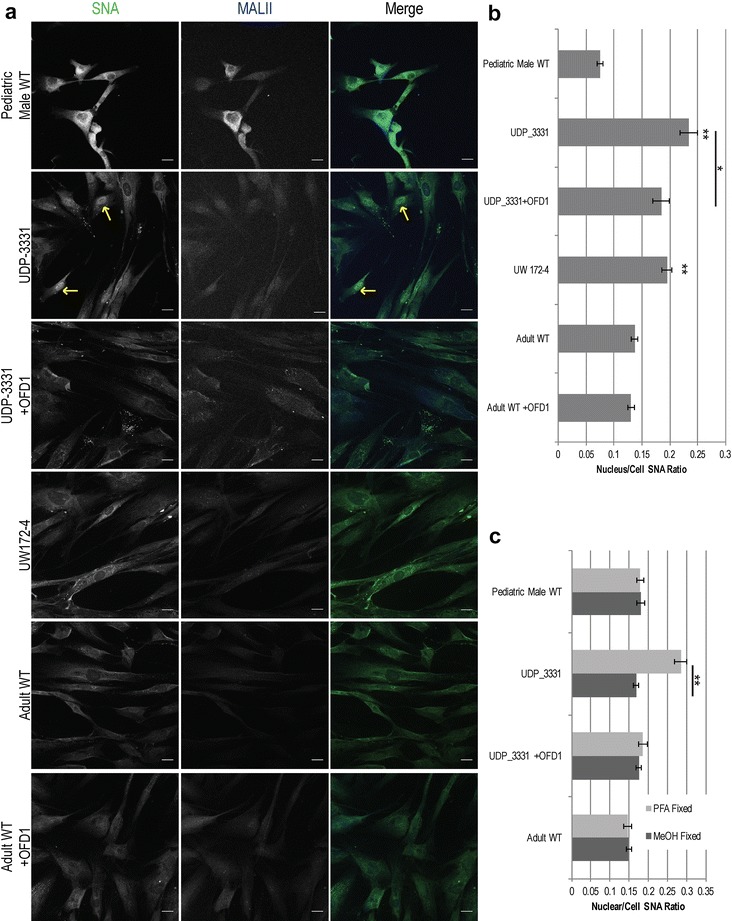
Results: Glycome analyses of plasma and cultured dermal fibroblasts identified abnormal N- and O-linked glycosylation profiles. These findings replicated in two unrelated males with OFD1 mutations. Cultured fibroblasts from affected individuals had a defect in ciliogenesis. The proband's fibroblasts also had an abnormally elevated nuclear sialylation signature and increased total cellular levels of CMP-sialic acid. Ciliogenesis and each glycosylation anomaly were rescued by expression of wild-type OFD1.
Conclusions: The rescue of ciliogenesis and glycosylation upon reintroduction of WT OFD1 suggests that both contribute to the pathogenesis of JBTS10.
Keywords: CMP-sialic acid; Ciliopathy; Glycosylation; Joubert syndrome; Molar tooth sign.
Figures




References
-
- Parisi M, Glass I. Joubert syndrome and related disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) Seattle: University of Washington; 1993. - PubMed
-
- Valente EM, Dallapiccola B, Bertini E. Joubert syndrome and related disorders. Handb Clin Neurol. 2013 - PubMed
-
- Webb TR, Parfitt DA, Gardner JC, Martinez A, Bevilacqua D, Davidson AE, Zito I, Thiselton DL, Ressa JH, Apergi M, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Hum Mol Genet. 2012 - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
