

INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions
- PMID: 28344988
- PMCID: PMC5363318
- DOI: 10.1016/j.omtm.2016.11.003
INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions
Abstract
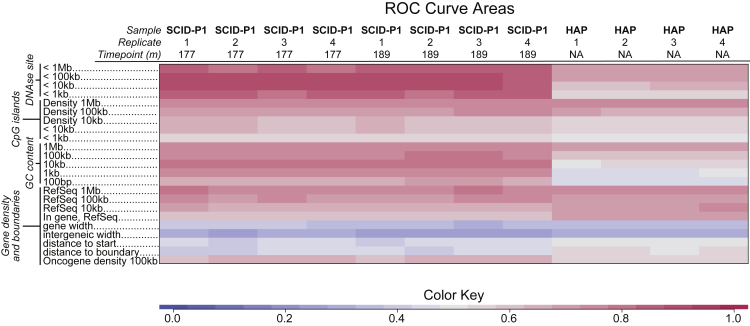
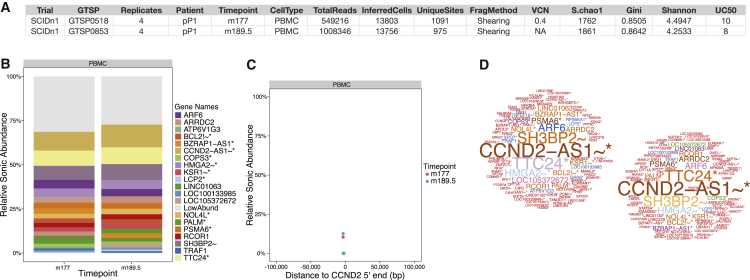
Analysis of sites of newly integrated DNA in cellular genomes is important to several fields, but methods for analyzing and visualizing these datasets are still under development. Here, we describe tools for data analysis and visualization that take as input integration site data from our INSPIIRED pipeline. Paired-end sequencing allows inference of the numbers of transduced cells as well as the distributions of integration sites in target genomes. We present interactive heatmaps that allow comparison of distributions of integration sites to genomic features and that support numerous user-defined statistical tests. To summarize integration site data from human gene therapy samples, we developed a reproducible report format that catalogs sample population structure, longitudinal dynamics, and integration frequency near cancer-associated genes. We also introduce a novel summary statistic, the UC50 (unique cell progenitors contributing the most expanded 50% of progeny cell clones), which provides a single number summarizing possible clonal expansion. Using these tools, we characterize ongoing longitudinal characterization of a patient from the first trial to treat severe combined immunodeficiency-X1 (SCID-X1), showing successful reconstitution for 15 years accompanied by persistence of a cell clone with an integration site near the cancer-associated gene CCND2. Software is available at https://github.com/BushmanLab/INSPIIRED.
Keywords: SCID-X1; data visualization; gammaretrovirus; gene therapy; insertional mutagenesis; lentivirus; mutagenesis; recombination; retrovirus; vector; vector driving.
Figures



References
-
- Bushman F.D. Cold Spring Harbor Laboratory Press; 2001. Lateral DNA Transfer: Mechanisms and Consequences.
-
- Craig N.L., Craigie R., Gellert M., Lambowitz A.M. American Society for Microbiology Press; Washington, D.C.: 2002. Mobile DNA II.
-
- Schröder A.R., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. - PubMed
-
- Coffin J.M., Hughes S.H., Varmus H.E. Cold Spring Harbor Laboratory Press; 1997. Retroviruses. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
