

Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production
- PMID: 28346360
- PMCID: PMC5412292
- DOI: 10.3390/ijms18040706
Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production
Abstract
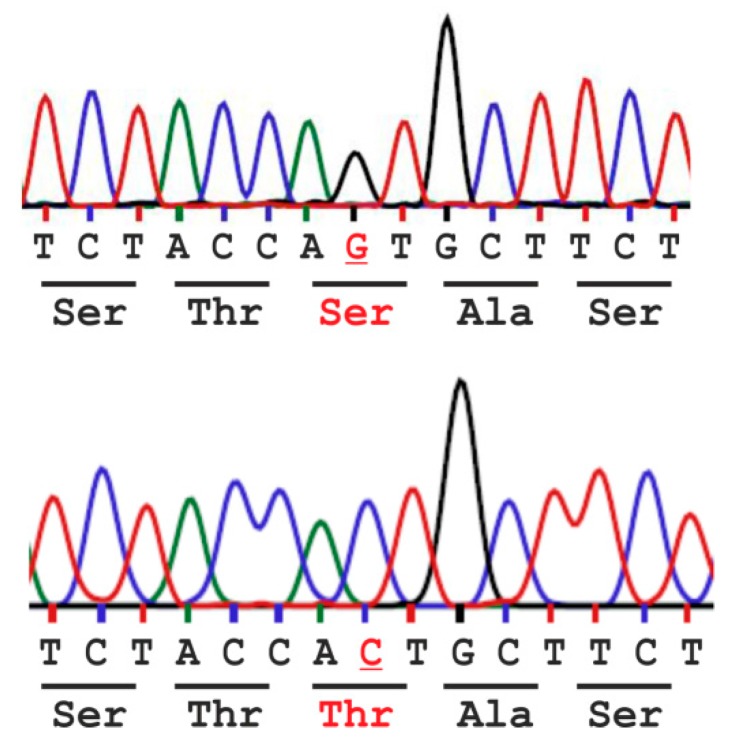
Aspartylglucosaminidase (AGA) is a lysosomal hydrolase that participates in the breakdown of glycoproteins. Defects in the AGA gene result in a lysosomal storage disorder, aspartylglucosaminuria (AGU), that manifests mainly as progressive mental retardation. A number of AGU missense mutations have been identified that result in reduced AGA activity. Human variants that contain either Ser or Thr in position 149 have been described, but it is unknown if this affects AGA processing or activity. Here, we have directly compared the Ser149/Thr149 variants of AGA and show that they do not differ in terms of relative specific activity or processing. Therefore, Thr149 AGA, which is the rare variant, can be considered as a neutral or benign variant. Furthermore, we have here produced codon-optimized versions of these two variants and show that they are expressed at significantly higher levels than AGA with the natural codon-usage. Since optimal AGA expression is of vital importance for both gene therapy and enzyme replacement, our data suggest that use of codon-optimized AGA may be beneficial for these therapy options.
Keywords: aspartylglucosaminidase; aspartylglucosaminuria; gene therapy; lysosomal storage disorder; lysosomes; single nucleotide polymorphism.
Conflict of interest statement
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Figures





Similar articles
-
Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria.Biochim Biophys Acta Mol Basis Dis. 2018 Mar;1864(3):668-675. doi: 10.1016/j.bbadis.2017.12.014. Epub 2017 Dec 13. Biochim Biophys Acta Mol Basis Dis. 2018. PMID: 29247835
-
Adenovirus-mediated gene transfer results in decreased lysosomal storage in brain and total correction in liver of aspartylglucosaminuria (AGU) mouse.Gene Ther. 1998 Oct;5(10):1314-21. doi: 10.1038/sj.gt.3300740. Gene Ther. 1998. PMID: 9930336
-
Molecular pathogenesis of a disease: structural consequences of aspartylglucosaminuria mutations.Hum Mol Genet. 2001 Apr 15;10(9):983-95. doi: 10.1093/hmg/10.9.983. Hum Mol Genet. 2001. PMID: 11309371
-
Aspartylglycosaminuria: a review.Orphanet J Rare Dis. 2016 Dec 1;11(1):162. doi: 10.1186/s13023-016-0544-6. Orphanet J Rare Dis. 2016. PMID: 27906067 Free PMC article. Review.
-
Aspartylglycosaminuria: biochemistry and molecular biology.Biochim Biophys Acta. 1999 Oct 8;1455(2-3):139-54. doi: 10.1016/s0925-4439(99)00076-9. Biochim Biophys Acta. 1999. PMID: 10571008 Review.
Cited by
-
Pre-clinical Gene Therapy with AAV9/AGA in Aspartylglucosaminuria Mice Provides Evidence for Clinical Translation.Mol Ther. 2021 Mar 3;29(3):989-1000. doi: 10.1016/j.ymthe.2020.11.012. Epub 2020 Nov 11. Mol Ther. 2021. PMID: 33186692 Free PMC article.
-
The T99K variant of glycosylasparaginase shows a new structural mechanism of the genetic disease aspartylglucosaminuria.Protein Sci. 2019 Jun;28(6):1013-1023. doi: 10.1002/pro.3607. Epub 2019 Apr 9. Protein Sci. 2019. PMID: 30901125 Free PMC article.
-
Validation of Aspartylglucosaminidase Activity Assay for Human Serum Samples: Establishment of a Biomarker for Diagnostics and Clinical Studies.Int J Mol Sci. 2023 Mar 16;24(6):5722. doi: 10.3390/ijms24065722. Int J Mol Sci. 2023. PMID: 36982794 Free PMC article.
-
Development of a Lentiviral Vector for High-Yield Production of Synthetic and Recombinant GCase for Gaucher Disease Therapy.Int J Mol Sci. 2025 Jul 23;26(15):7089. doi: 10.3390/ijms26157089. Int J Mol Sci. 2025. PMID: 40806226 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
