

The Process and Regulatory Components of Inflammation in Brain Oncogenesis
- PMID: 28346397
- PMCID: PMC5485723
- DOI: 10.3390/biom7020034
The Process and Regulatory Components of Inflammation in Brain Oncogenesis
Abstract
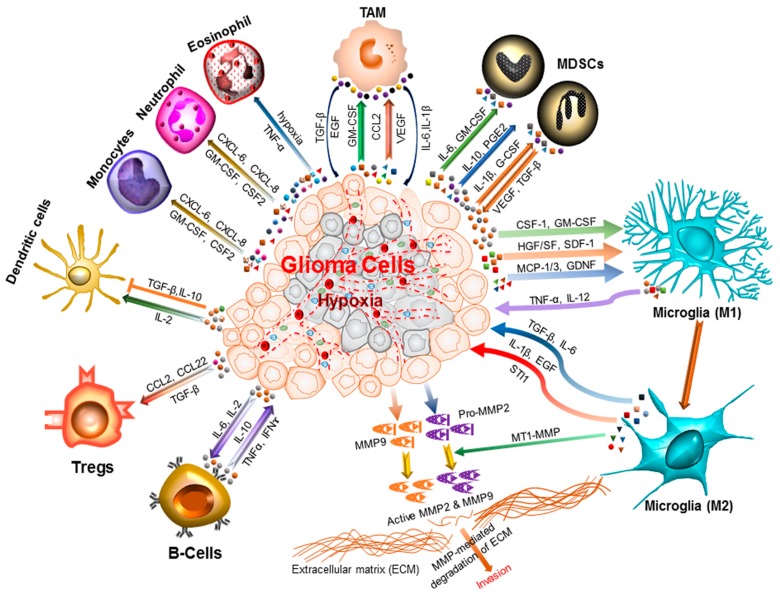
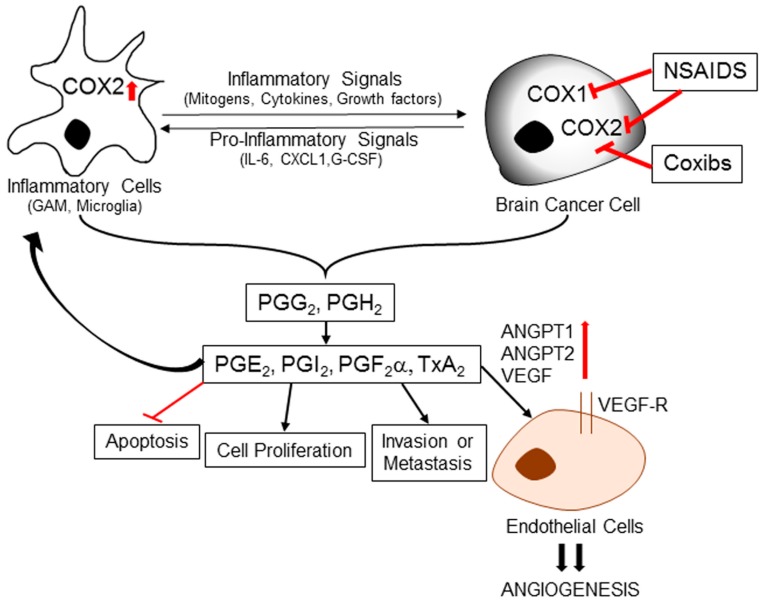
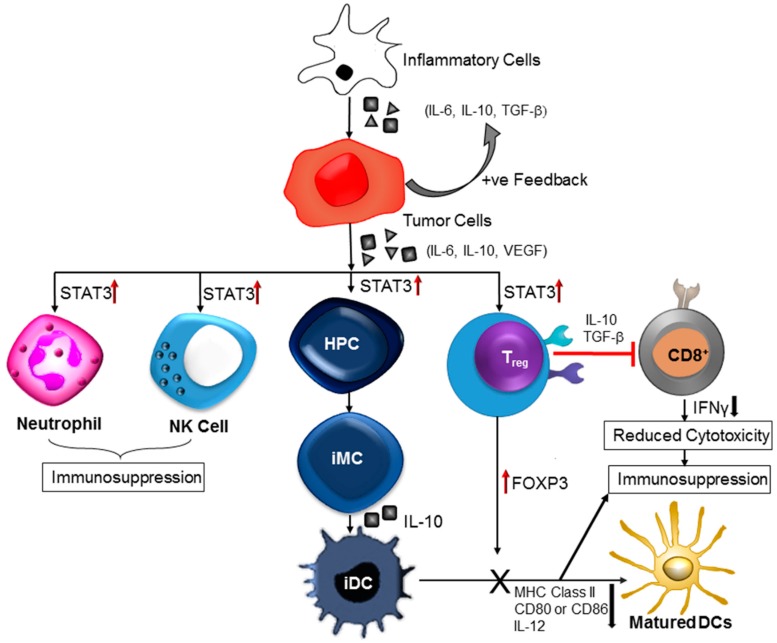
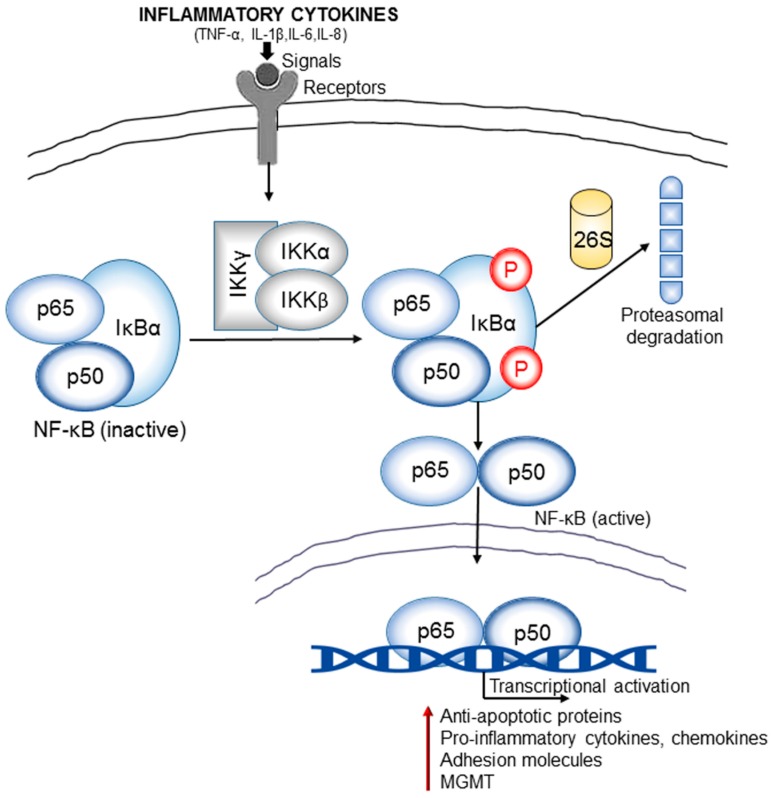
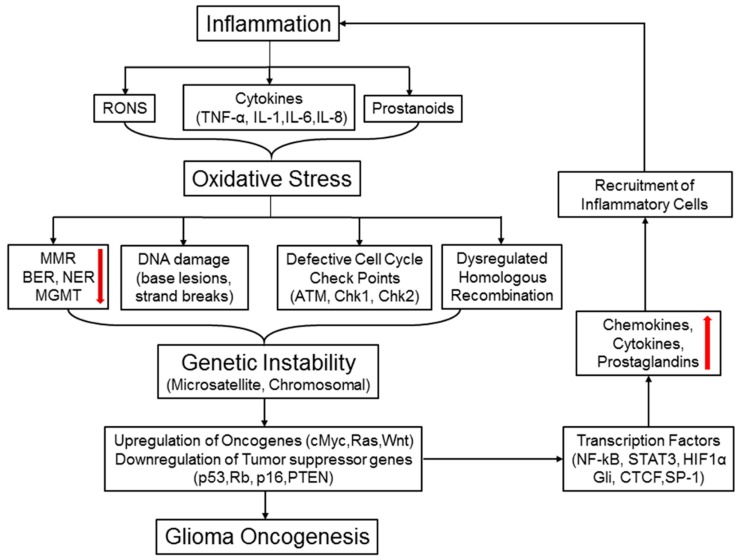
Central nervous system tumors comprising the primary cancers and brain metastases remain the most lethal neoplasms and challenging to treat. Substantial evidence points to a paramount role for inflammation in the pathology leading to gliomagenesis, malignant progression and tumor aggressiveness in the central nervous system (CNS) microenvironment. This review summarizes the salient contributions of oxidative stress, interleukins, tumor necrosis factor-α (TNF-α), cyclooxygenases, and transcription factors such as signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) and the associated cross-talks to the inflammatory signaling in CNS cancers. The roles of reactive astrocytes, tumor associated microglia and macrophages, metabolic alterations, microsatellite instability, O⁶-methylguanine DNA methyltransferase (MGMT) DNA repair and epigenetic alterations mediated by the isocitrate dehydrogenase 1 (IDH1) mutations have been discussed. The inflammatory pathways with relevance to the brain cancer treatments have been highlighted.
Keywords: MGMT DNA repair; STAT3; epigenetic deregulation; gliomas; inflammation; interleukins.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- PDQ Adult Treatment Editorial Board . PDQ Cancer Information Summaries. National Cancer Institute; Bethesda, MD, USA: 2002. Adult Treatment Editorial Board. Adult central nervous system tumors treatment (PDQ®): Health professional version. - PubMed
-
- Berghoff A.S., Preusser M. The inflammatory microenvironment in brain metastases: Potential treatment target? Chin. Clin. Oncol. 2015;4:21. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
