

Near-infrared optogenetic pair for protein regulation and spectral multiplexing
- PMID: 28346403
- PMCID: PMC6239862
- DOI: 10.1038/nchembio.2343
Near-infrared optogenetic pair for protein regulation and spectral multiplexing
Abstract
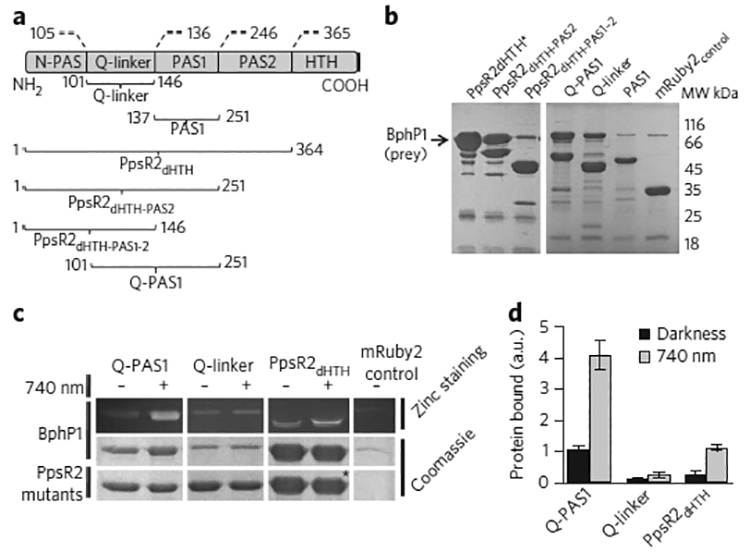
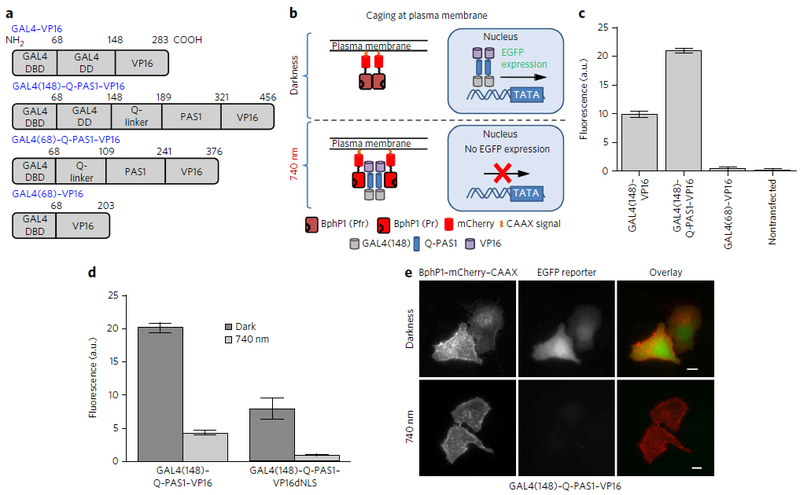
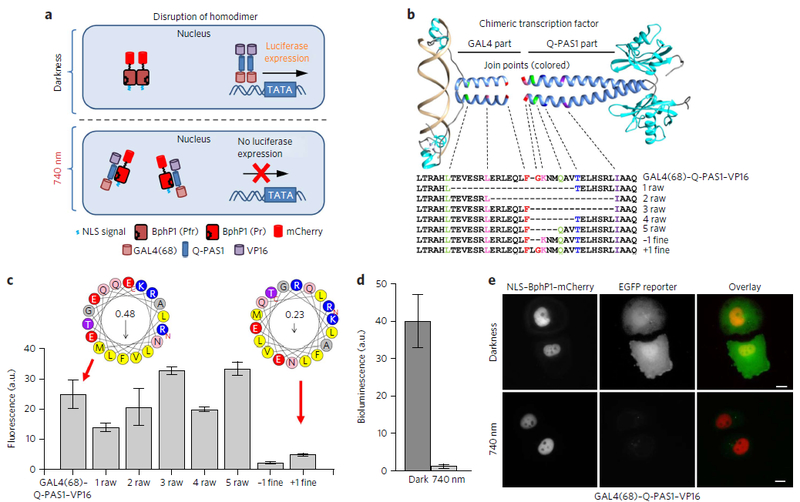
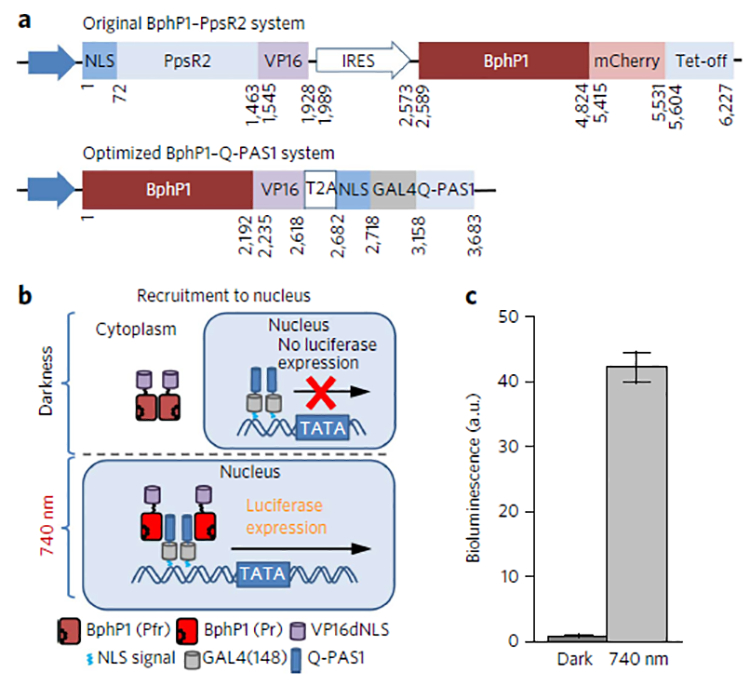
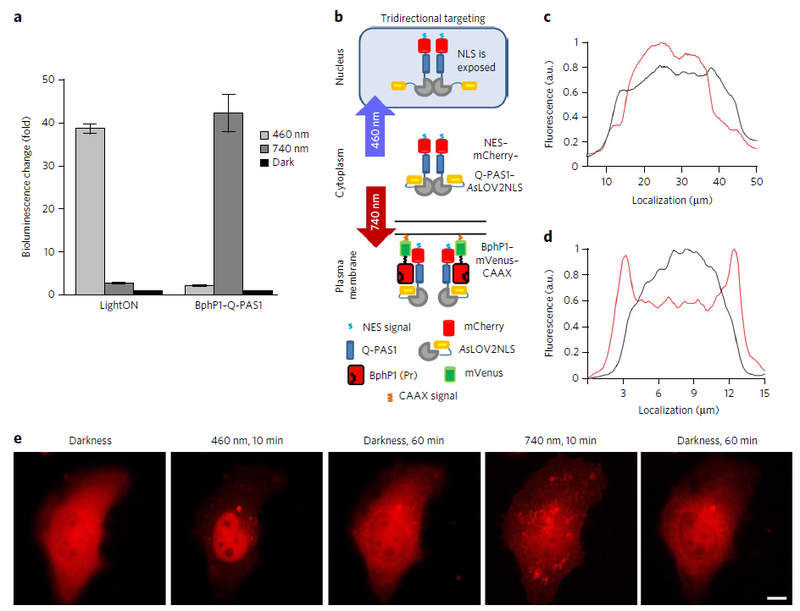
Multifunctional optogenetic systems are in high demand for use in basic and biomedical research. Near-infrared-light-inducible binding of bacterial phytochrome BphP1 to its natural PpsR2 partner is beneficial for simultaneous use with blue-light-activatable tools. However, applications of the BphP1-PpsR2 pair are limited by the large size, multidomain structure and oligomeric behavior of PpsR2. Here, we engineered a single-domain BphP1 binding partner, Q-PAS1, which is three-fold smaller and lacks oligomerization. We exploited a helix-PAS fold of Q-PAS1 to develop several near-infrared-light-controllable transcription regulation systems, enabling either 40-fold activation or inhibition. The light-induced BphP1-Q-PAS1 interaction allowed modification of the chromatin epigenetic state. Multiplexing the BphP1-Q-PAS1 pair with a blue-light-activatable LOV-domain-based system demonstrated their negligible spectral crosstalk. By integrating the Q-PAS1 and LOV domains in a single optogenetic tool, we achieved tridirectional protein targeting, independently controlled by near-infrared and blue light, thus demonstrating the superiority of Q-PAS1 for spectral multiplexing and engineering of multicomponent systems.
Conflict of interest statement
Figures
Comment in
-
Optogenetics: Switching with red and blue.Nat Chem Biol. 2017 May 17;13(6):573-574. doi: 10.1038/nchembio.2387. Nat Chem Biol. 2017. PMID: 28514429 No abstract available.
-
Optogenetic tools for cell biological applications.J Thorac Dis. 2017 Dec;9(12):4867-4870. doi: 10.21037/jtd.2017.11.73. J Thorac Dis. 2017. PMID: 29312679 Free PMC article. No abstract available.
References
-
- Wang X, Chen X & Yang Y Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Methods 9, 266–269(2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
