

Overlapping SETBP1 gain-of-function mutations in Schinzel-Giedion syndrome and hematologic malignancies
- PMID: 28346496
- PMCID: PMC5386295
- DOI: 10.1371/journal.pgen.1006683
Overlapping SETBP1 gain-of-function mutations in Schinzel-Giedion syndrome and hematologic malignancies
Abstract
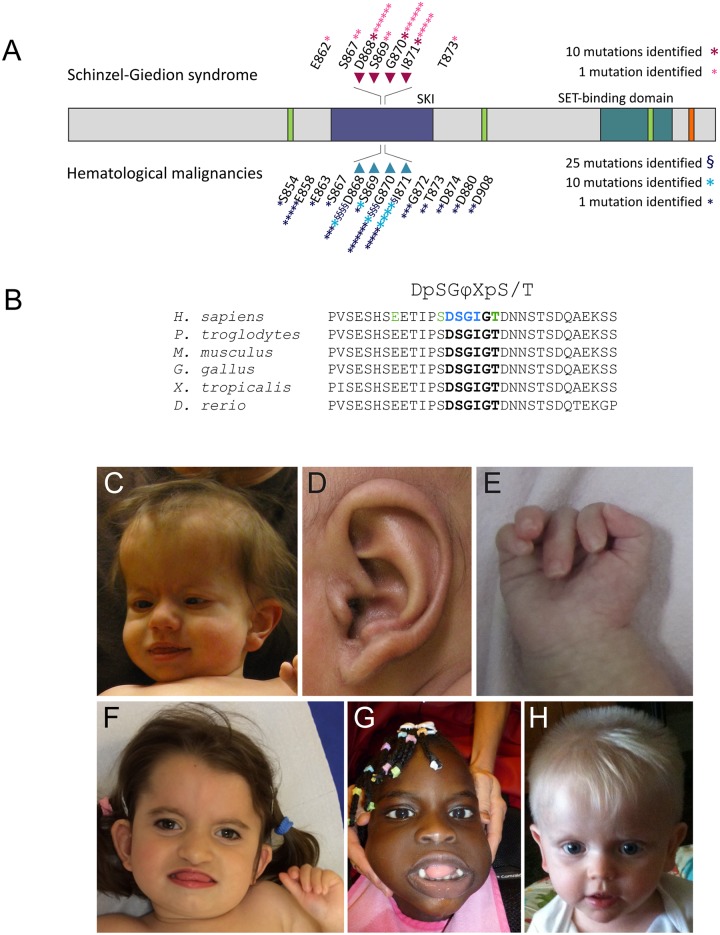
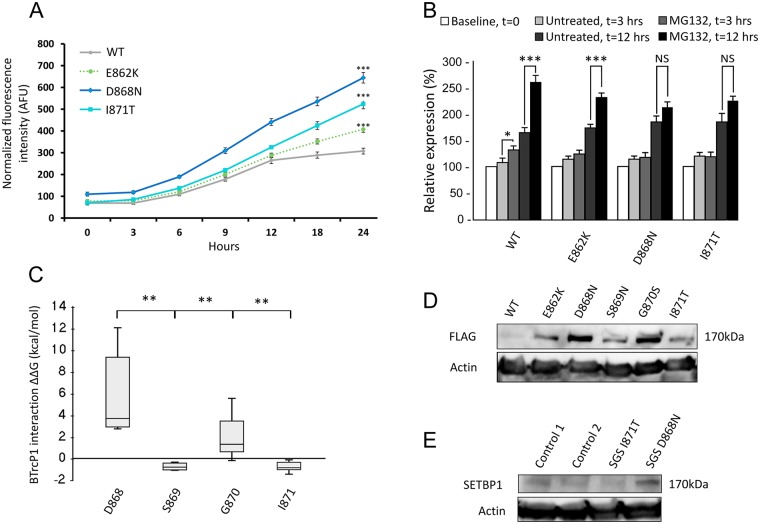
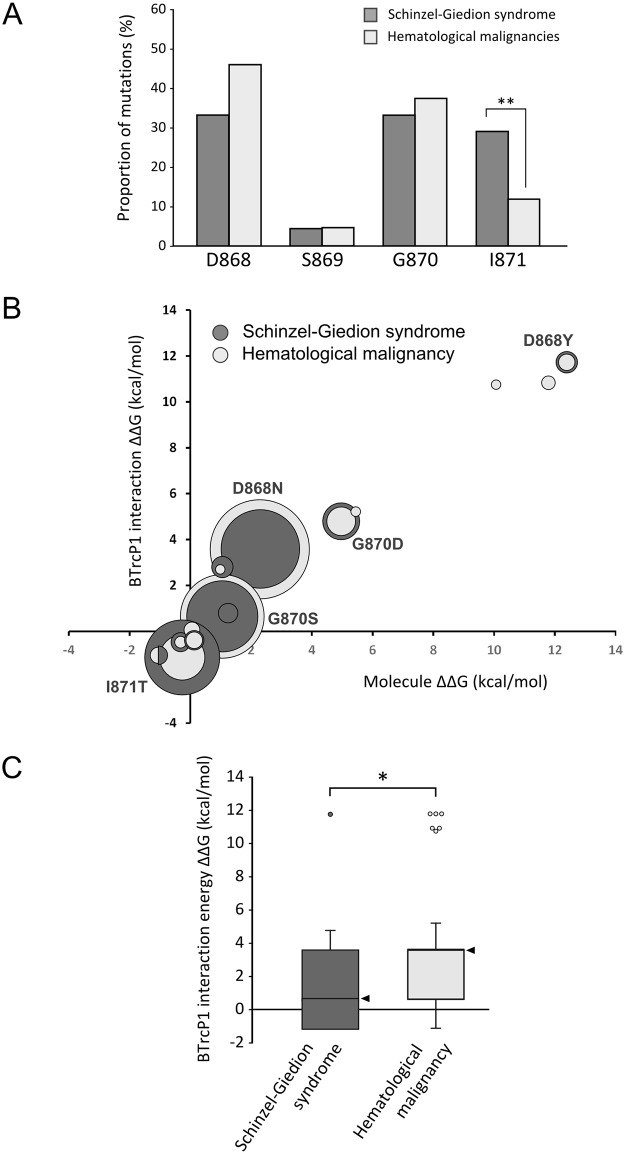
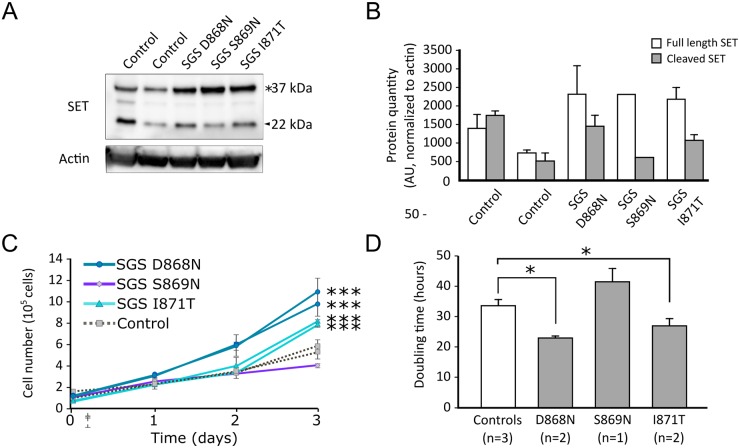
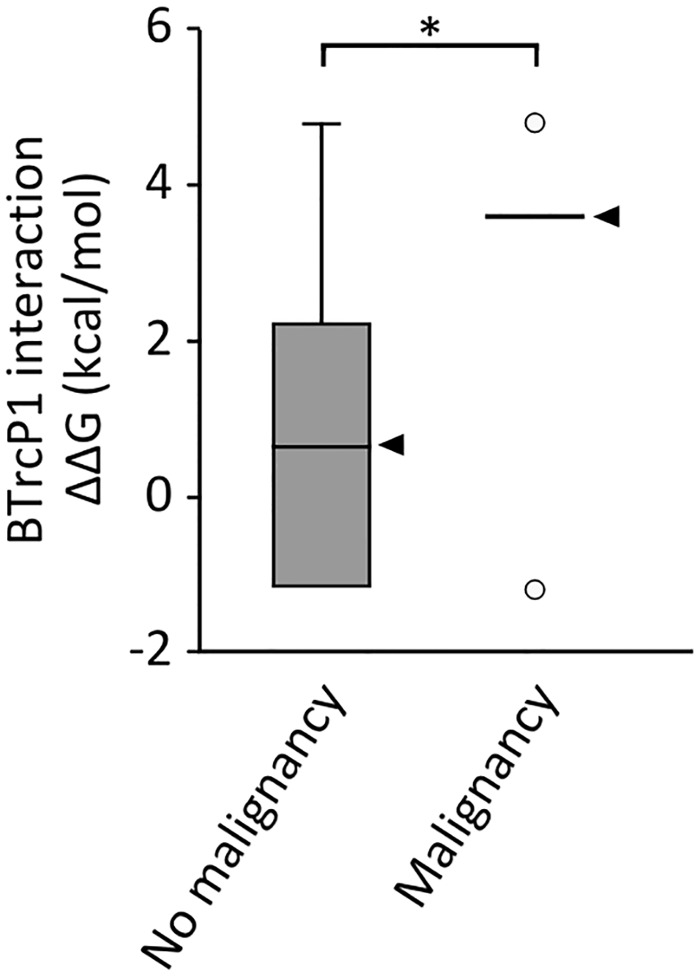
Schinzel-Giedion syndrome (SGS) is a rare developmental disorder characterized by multiple malformations, severe neurological alterations and increased risk of malignancy. SGS is caused by de novo germline mutations clustering to a 12bp hotspot in exon 4 of SETBP1. Mutations in this hotspot disrupt a degron, a signal for the regulation of protein degradation, and lead to the accumulation of SETBP1 protein. Overlapping SETBP1 hotspot mutations have been observed recurrently as somatic events in leukemia. We collected clinical information of 47 SGS patients (including 26 novel cases) with germline SETBP1 mutations and of four individuals with a milder phenotype caused by de novo germline mutations adjacent to the SETBP1 hotspot. Different mutations within and around the SETBP1 hotspot have varying effects on SETBP1 stability and protein levels in vitro and in in silico modeling. Substitutions in SETBP1 residue I871 result in a weak increase in protein levels and mutations affecting this residue are significantly more frequent in SGS than in leukemia. On the other hand, substitutions in residue D868 lead to the largest increase in protein levels. Individuals with germline mutations affecting D868 have enhanced cell proliferation in vitro and higher incidence of cancer compared to patients with other germline SETBP1 mutations. Our findings substantiate that, despite their overlap, somatic SETBP1 mutations driving malignancy are more disruptive to the degron than germline SETBP1 mutations causing SGS. Additionally, this suggests that the functional threshold for the development of cancer driven by the disruption of the SETBP1 degron is higher than for the alteration in prenatal development in SGS. Drawing on previous studies of somatic SETBP1 mutations in leukemia, our results reveal a genotype-phenotype correlation in germline SETBP1 mutations spanning a molecular, cellular and clinical phenotype.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
