

Specific expression of novel long non-coding RNAs in high-hyperdiploid childhood acute lymphoblastic leukemia
- PMID: 28346506
- PMCID: PMC5367703
- DOI: 10.1371/journal.pone.0174124
Specific expression of novel long non-coding RNAs in high-hyperdiploid childhood acute lymphoblastic leukemia
Abstract
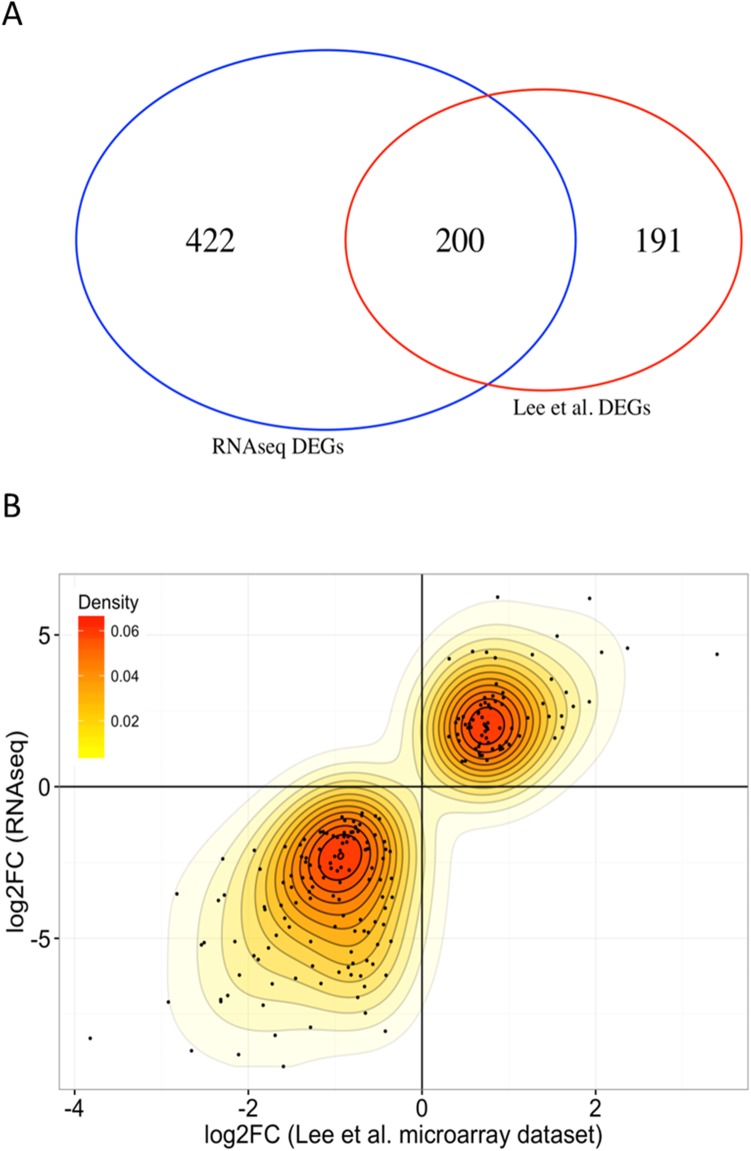
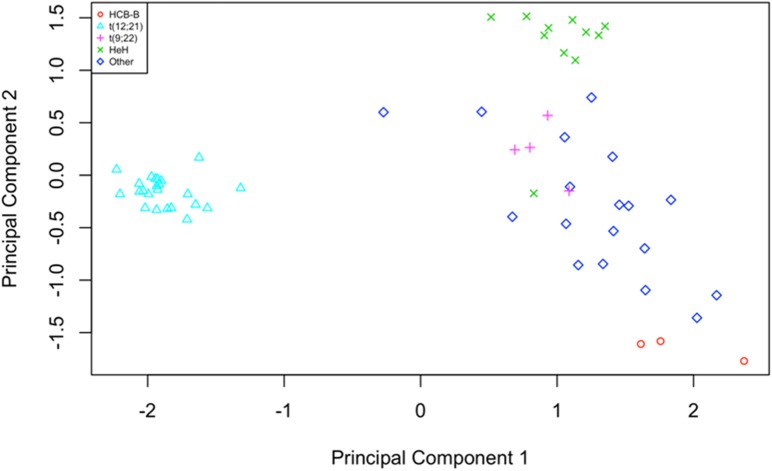
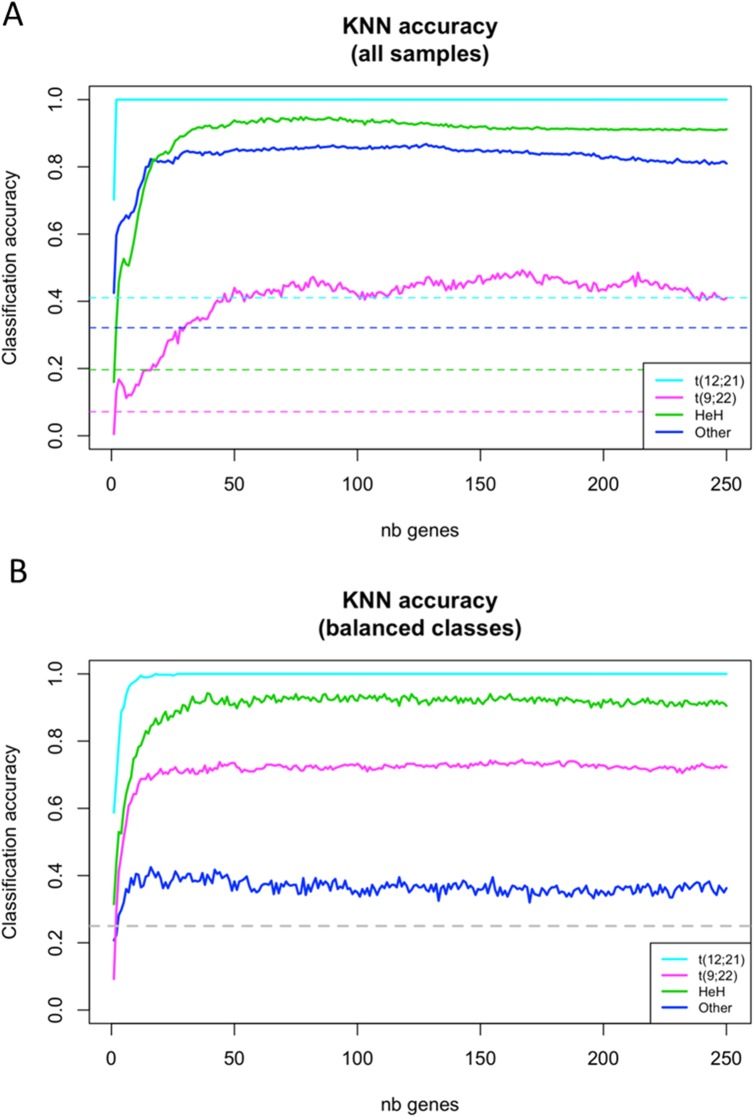
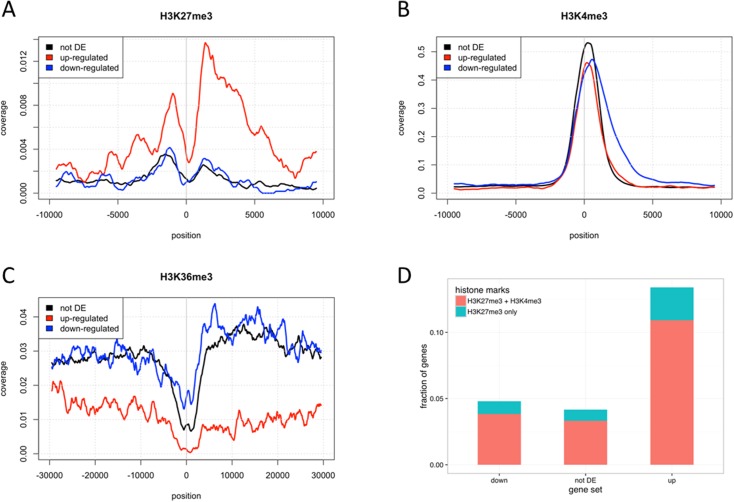
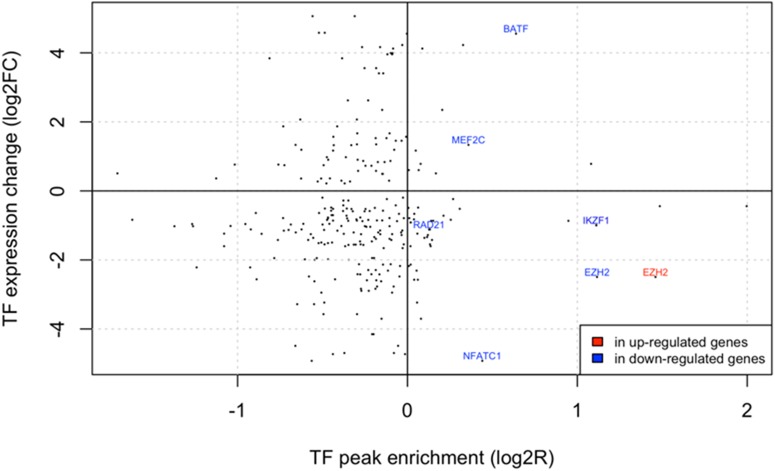
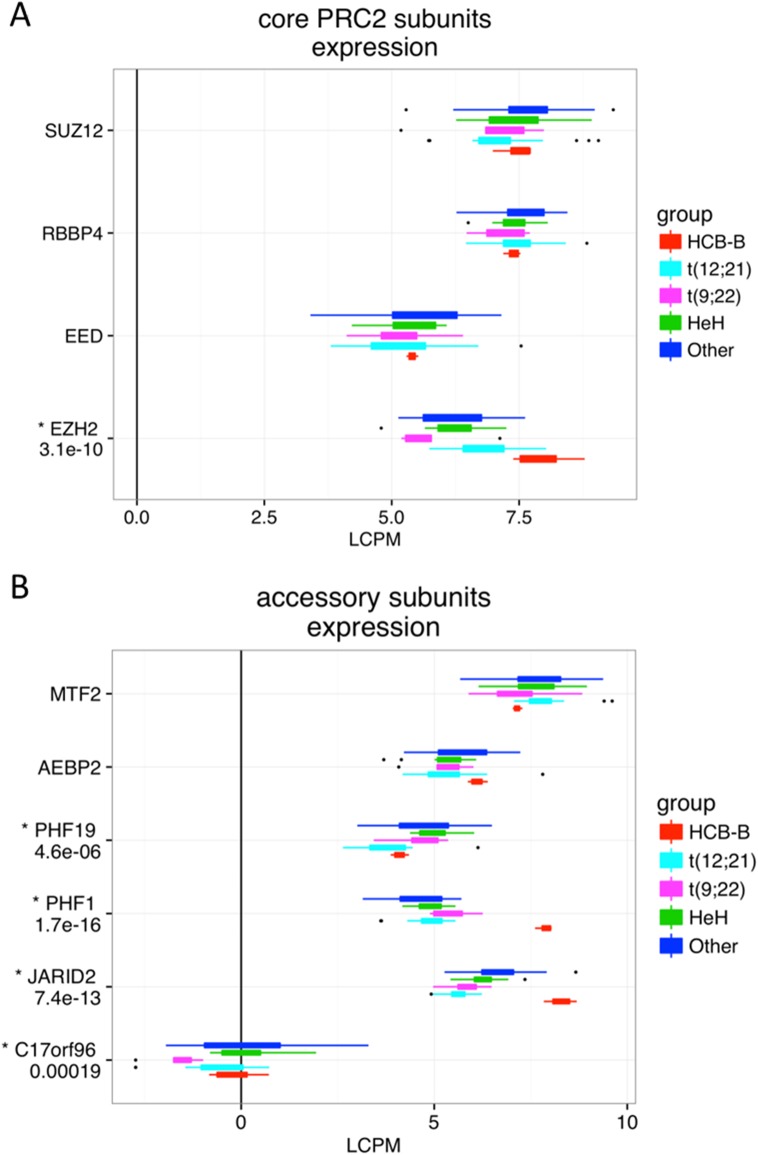
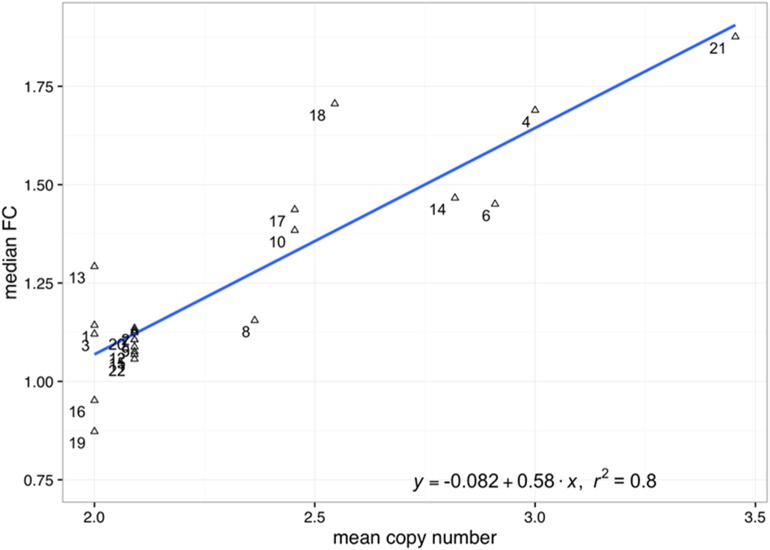
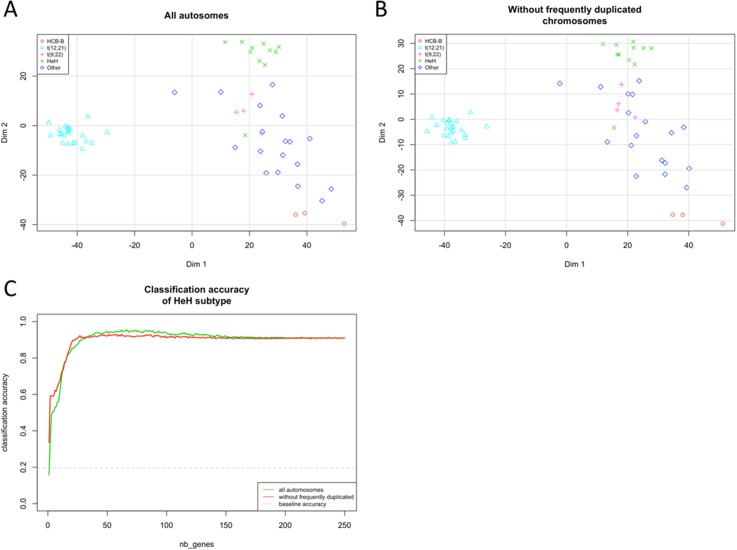
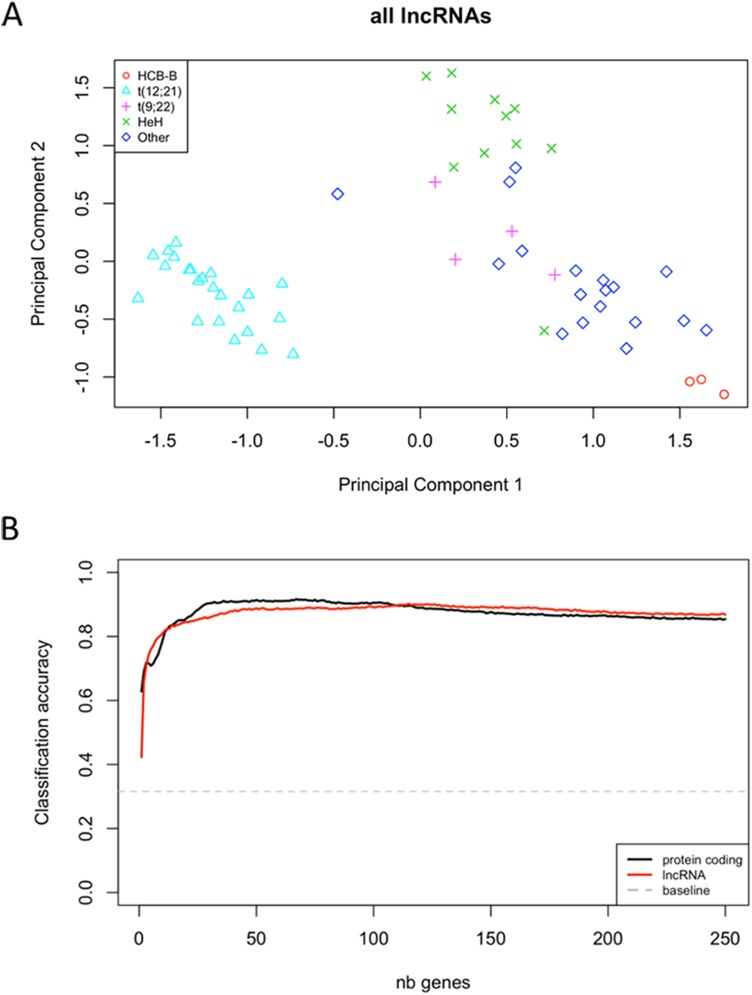
Pre-B cell childhood acute lymphoblastic leukemia (pre-B cALL) is a heterogeneous disease involving many subtypes typically stratified using a combination of cytogenetic and molecular-based assays. These methods, although widely used, rely on the presence of known chromosomal translocations, which is a limiting factor. There is therefore a need for robust, sensitive, and specific molecular biomarkers unaffected by such limitations that would allow better risk stratification and consequently better clinical outcome. In this study we performed a transcriptome analysis of 56 pre-B cALL patients to identify expression signatures in different subtypes. In both protein-coding and long non-coding RNAs (lncRNA), we identified subtype-specific gene signatures distinguishing pre-B cALL subtypes, particularly in t(12;21) and hyperdiploid cases. The genes up-regulated in pre-B cALL subtypes were enriched in bivalent chromatin marks in their promoters. LncRNAs is a new and under-studied class of transcripts. The subtype-specific nature of lncRNAs suggests they may be suitable clinical biomarkers to guide risk stratification and targeted therapies in pre-B cALL patients.
Conflict of interest statement
Figures
References
-
- Kaneko Y, Hayashi Y, Sakurai M (1981) Chromosomal findings and their correlation to prognosis in acute lymphocytic leukemia. Cancer Genet Cytogenet 4: 227–235. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
