

Inhibition of WNT signaling in the bone marrow niche prevents the development of MDS in the Apcdel/+ MDS mouse model
- PMID: 28348148
- PMCID: PMC5454335
- DOI: 10.1182/blood-2016-08-736454
Inhibition of WNT signaling in the bone marrow niche prevents the development of MDS in the Apcdel/+ MDS mouse model
Abstract
There is accumulating evidence that functional alteration(s) of the bone marrow (BM) microenvironment contribute to the development of some myeloid disorders, such as myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). In addition to a cell-intrinsic role of WNT activation in leukemia stem cells, WNT activation in the BM niche is also thought to contribute to the pathogenesis of MDS and AML. We previously showed that the Apc-haploinsufficient mice (Apcdel/+ ) model MDS induced by an aberrant BM microenvironment. We sought to determine whether Apc, a multifunctional protein and key negative regulator of the canonical β-catenin (Ctnnb1)/WNT-signaling pathway, mediates this disease through modulating WNT signaling, and whether inhibition of WNT signaling prevents the development of MDS in Apcdel/+ mice. Here, we demonstrate that loss of 1 copy of Ctnnb1 is sufficient to prevent the development of MDS in Apcdel/+ mice and that altered canonical WNT signaling in the microenvironment is responsible for the disease. Furthermore, the US Food and Drug Administration (FDA)-approved drug pyrvinium delays and/or inhibits disease in Apcdel/+ mice, even when it is administered after the presentation of anemia. Other groups have observed increased nuclear CTNNB1 in stromal cells from a high frequency of MDS/AML patients, a finding that together with our results highlights a potential new strategy for treating some myeloid disorders.
© 2017 by The American Society of Hematology.
Figures
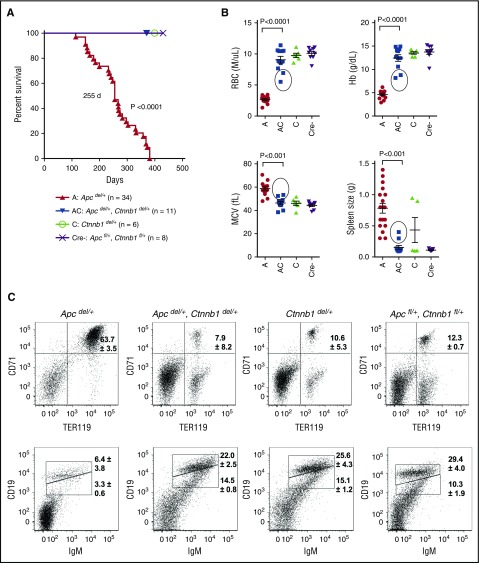
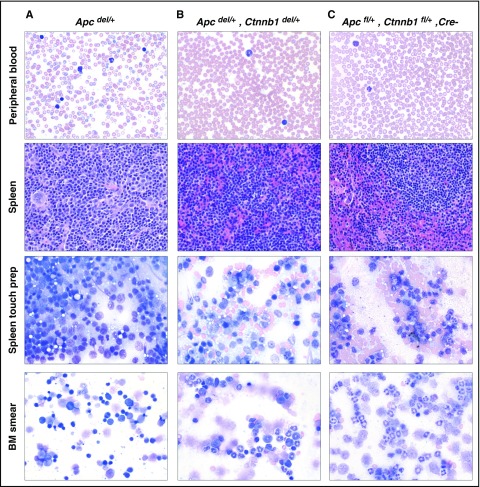
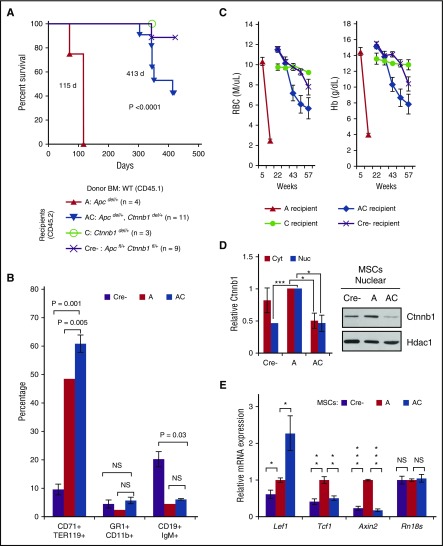
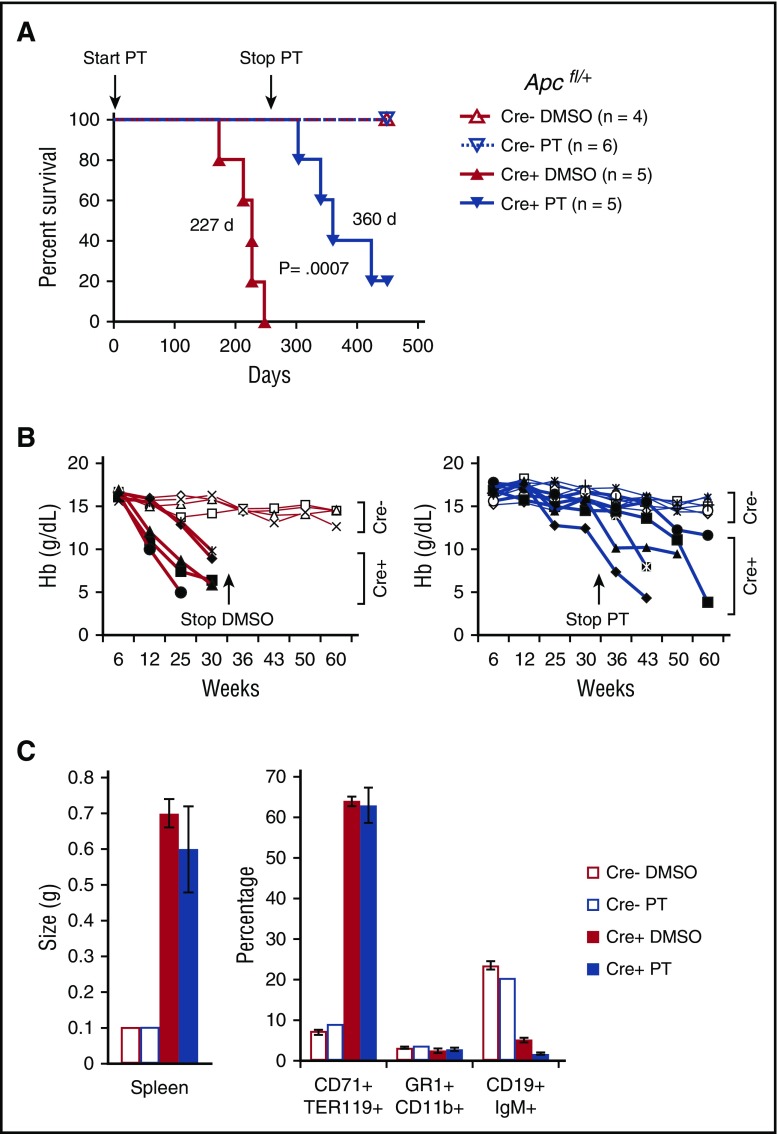
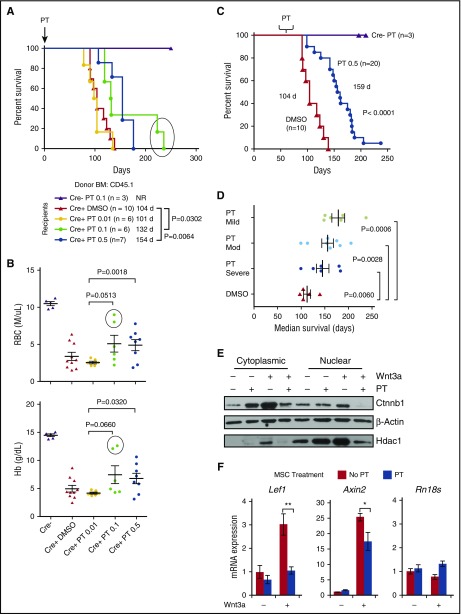
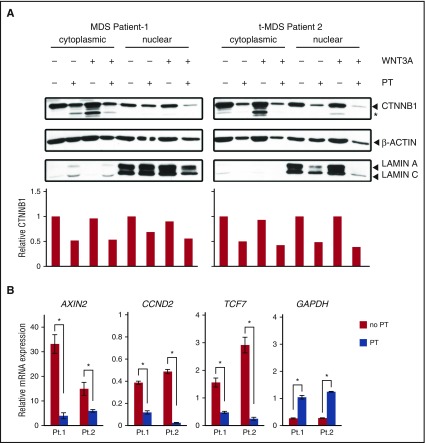
Comment in
-
Hacking the stem cell niche.Blood. 2017 Jun 1;129(22):2951-2952. doi: 10.1182/blood-2017-04-777789. Blood. 2017. PMID: 28572172 No abstract available.
Similar articles
-
β-Catenin Is a Candidate Therapeutic Target for Myeloid Neoplasms with del(5q).Cancer Res. 2017 Aug 1;77(15):4116-4126. doi: 10.1158/0008-5472.CAN-17-0202. Epub 2017 Jun 13. Cancer Res. 2017. PMID: 28611040 Free PMC article.
-
The Apc(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS.Blood. 2010 Apr 29;115(17):3489-97. doi: 10.1182/blood-2009-11-251728. Epub 2010 Mar 2. Blood. 2010. PMID: 20197553 Free PMC article.
-
Impairment of PI3K/AKT and WNT/β-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes.Exp Hematol. 2016 Jan;44(1):75-83.e1-4. doi: 10.1016/j.exphem.2015.10.005. Epub 2015 Oct 28. Exp Hematol. 2016. PMID: 26521017
-
Bone marrow niche in the myelodysplastic syndromes.Leuk Res. 2015 Oct;39(10):1020-7. doi: 10.1016/j.leukres.2015.06.017. Epub 2015 Jul 14. Leuk Res. 2015. PMID: 26276090 Review.
-
Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis-Masters of Survival and Clonality?Int J Mol Sci. 2016 Jun 27;17(7):1009. doi: 10.3390/ijms17071009. Int J Mol Sci. 2016. PMID: 27355944 Free PMC article. Review.
Cited by
-
Ryk modulates the niche activity of mesenchymal stromal cells by fine-tuning canonical Wnt signaling.Exp Mol Med. 2020 Jul;52(7):1140-1151. doi: 10.1038/s12276-020-0477-y. Epub 2020 Jul 28. Exp Mol Med. 2020. PMID: 32724069 Free PMC article.
-
DNA damage to bone marrow stromal cells by antileukemia drugs induces chemoresistance in acute myeloid leukemia via paracrine FGF10-FGFR2 signaling.J Biol Chem. 2023 Jan;299(1):102787. doi: 10.1016/j.jbc.2022.102787. Epub 2022 Dec 9. J Biol Chem. 2023. PMID: 36509141 Free PMC article.
-
MSI2 mediates WNT/β-Catenin pathway function in hematopoietic stem cells.Leukemia. 2025 Jan;39(1):265-270. doi: 10.1038/s41375-024-02447-9. Epub 2024 Oct 22. Leukemia. 2025. PMID: 39438589 No abstract available.
-
The Bone's Role in Myeloid Neoplasia.Int J Mol Sci. 2020 Jul 1;21(13):4712. doi: 10.3390/ijms21134712. Int J Mol Sci. 2020. PMID: 32630305 Free PMC article. Review.
-
β-Catenin interacts with canonical RBPs including MSI2 to associate with a Wnt signalling mRNA network in myeloid leukaemia cells.Oncogene. 2025 Aug;44(29):2490-2503. doi: 10.1038/s41388-025-03415-y. Epub 2025 Apr 30. Oncogene. 2025. PMID: 40301545 Free PMC article.
References
-
- Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361(19):1872-1885. - PubMed
-
- Smith SM, Le Beau MM, Huo D, et al. . Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102(1):43-52. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
