

Recombination produces coherent bacterial species clusters in both core and accessory genomes
- PMID: 28348822
- PMCID: PMC5320679
- DOI: 10.1099/mgen.0.000038
Recombination produces coherent bacterial species clusters in both core and accessory genomes
Abstract
Background: Population samples show bacterial genomes can be divided into a core of ubiquitous genes and accessory genes that are present in a fraction of isolates. The ecological significance of this variation in gene content remains unclear. However, microbiologists agree that a bacterial species should be 'genomically coherent', even though there is no consensus on how this should be determined.
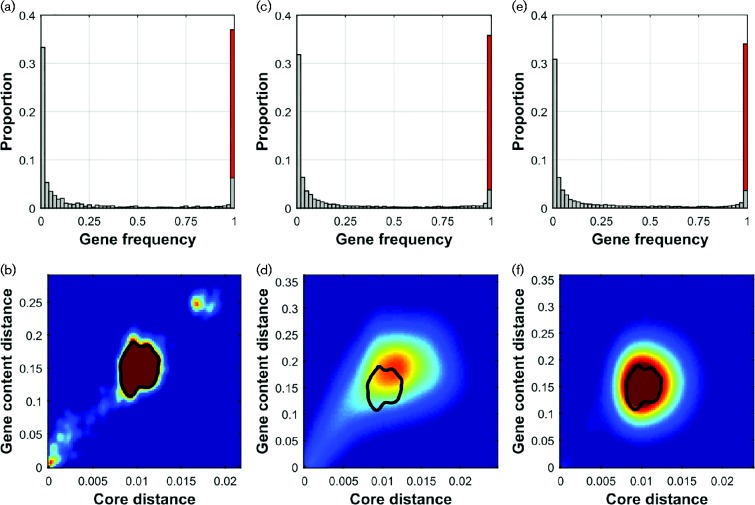
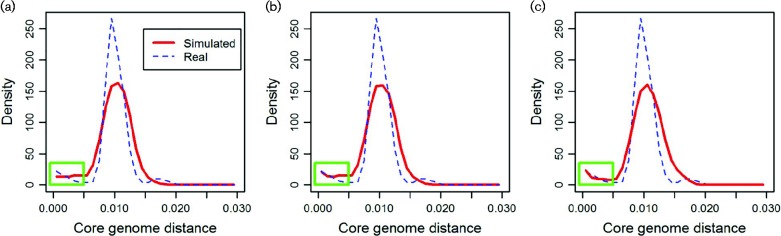
Results: We use a parsimonious model combining diversification in both the core and accessory genome, including mutation, homologous recombination (HR) and horizontal gene transfer (HGT) introducing new loci, to produce a population of interacting clusters of strains with varying genome content. New loci introduced by HGT may then be transferred on by HR. The model fits well to a systematic population sample of 616 pneumococcal genomes, capturing the major features of the population structure with parameter values that agree well with empirical estimates.
Conclusions: The model does not include explicit selection on individual genes, suggesting that crude comparisons of gene content may be a poor predictor of ecological function. We identify a clearly divergent subpopulation of pneumococci that are inconsistent with the model and may be considered genomically incoherent with the rest of the population. These strains have a distinct disease tropism and may be rationally defined as a separate species. We also find deviations from the model that may be explained by recent population bottlenecks or spatial structure.
Keywords: computational modeling; core/accessory genome; evolution; recombination; speciation.
Figures
References
Data Bibliography
-
- Marttinen, P., Croucher, N. J., Gutmann, M. U., Corander, J. & Hanage, W. P. (2015). Figshare. http://figshare.com/s/6471c982669011e58c4806ec4b8d1f61. - PMC - PubMed
-
- Marttinen, P., Croucher, N. J., Gutmann, M. U., Corander, J. & Hanage, W. P. (2015). Figshare. http://figshare.com/s/c70dd5e0669011e59ff906ec4bbcf141. - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources