

Evolutionary interpretations of mycobacteriophage biodiversity and host-range through the analysis of codon usage bias
- PMID: 28348827
- PMCID: PMC5359403
- DOI: 10.1099/mgen.0.000079
Evolutionary interpretations of mycobacteriophage biodiversity and host-range through the analysis of codon usage bias
Abstract
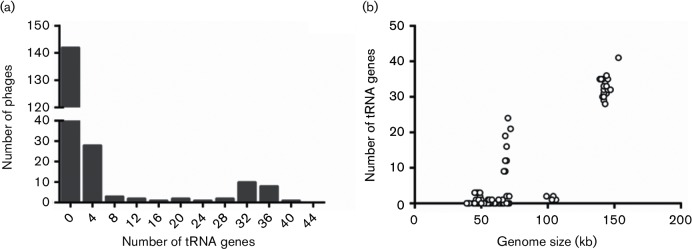
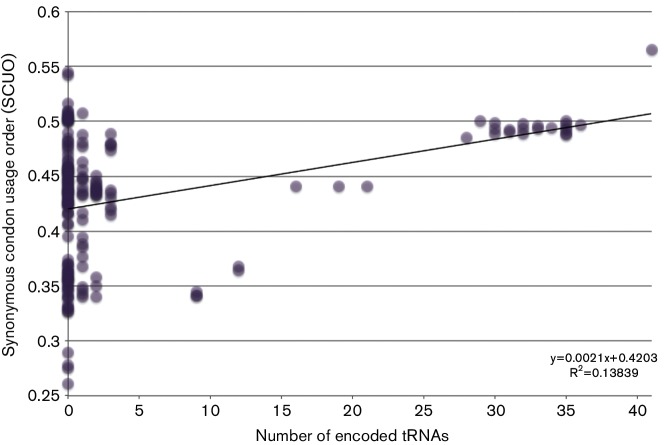
In an genomics course sponsored by the Howard Hughes Medical Institute (HHMI), undergraduate students have isolated and sequenced the genomes of more than 1,150 mycobacteriophages, creating the largest database of sequenced bacteriophages able to infect a single host, Mycobacterium smegmatis, a soil bacterium. Genomic analysis indicates that these mycobacteriophages can be grouped into 26 clusters based on genetic similarity. These clusters span a continuum of genetic diversity, with extensive genomic mosaicism among phages in different clusters. However, little is known regarding the primary hosts of these mycobacteriophages in their natural habitats, nor of their broader host ranges. As such, it is possible that the primary host of many newly isolated mycobacteriophages is not M. smegmatis, but instead a range of closely related bacterial species. However, determining mycobacteriophage host range presents difficulties associated with mycobacterial cultivability, pathogenicity and growth. Another way to gain insight into mycobacteriophage host range and ecology is through bioinformatic analysis of their genomic sequences. To this end, we examined the correlations between the codon usage biases of 199 different mycobacteriophages and those of several fully sequenced mycobacterial species in order to gain insight into the natural host range of these mycobacteriophages. We find that UPGMA clustering tends to match, but not consistently, clustering by shared nucleotide sequence identify. In addition, analysis of GC content, tRNA usage and correlations between mycobacteriophage and mycobacterial codon usage bias suggests that the preferred host of many clustered mycobacteriophages is not M. smegmatis but other, as yet unknown, members of the mycobacteria complex or closely allied bacterial species.
Keywords: UPGMA clustering; bacteriophages; codon bias; host range; mycobacteria; viral tRNAs.
Figures

References
Data Bibliography
-
- Alferez, G. I., Bryan, W. J., Byington, E. L., & other authors . NCBI GenBank, http://www.ncbi.nlm.nih.gov/nuccore/JF704105 - GenBank Accession #: JF70...
-
- Bambawale, V., Bieberich, J. C., Borowski, A. L., & other authors . NCBI GenBank, http://www.ncbi.nlm.nih.gov/nuccore/JF704116 - GenBank Accession #: JF70...
-
- Cole, S. T., Brosch, R., Parkhill, J., & other authors. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537-544. GenBank Accession #: NC_000962 (2001). - PubMed
-
- Cole, S. T., Eiglmeier, K., Parkhill, J., & other authors. Massive gene decay in the leprosy bacillus. Nature 409, 1007-1011. GenBank Accession #: NC_002677 (2001). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous