

Positional bias in variant calls against draft reference assemblies
- PMID: 28351369
- PMCID: PMC5368935
- DOI: 10.1186/s12864-017-3637-2
Positional bias in variant calls against draft reference assemblies
Abstract
Background: Whole genome resequencing projects may implement variant calling using draft reference genomes assembled de novo from short-read libraries. Despite lower quality of such assemblies, they allowed researchers to extend a wide range of population genetic and genome-wide association analyses to non-model species. As the variant calling pipelines are complex and involve many software packages, it is important to understand inherent biases and limitations at each step of the analysis.
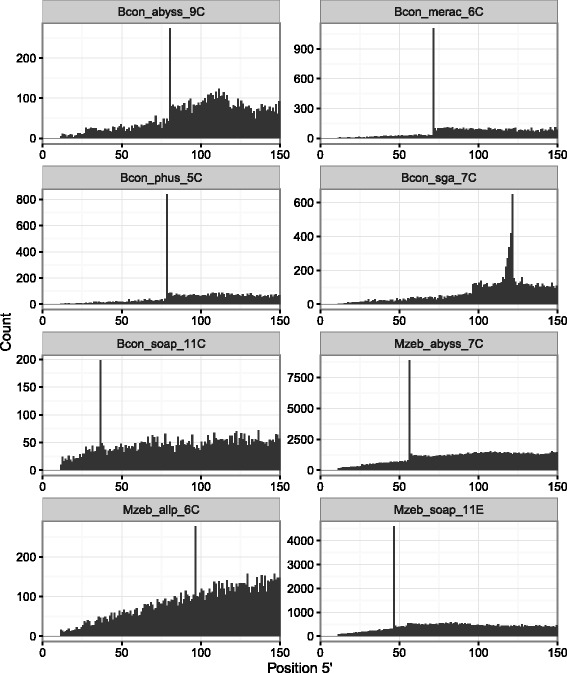
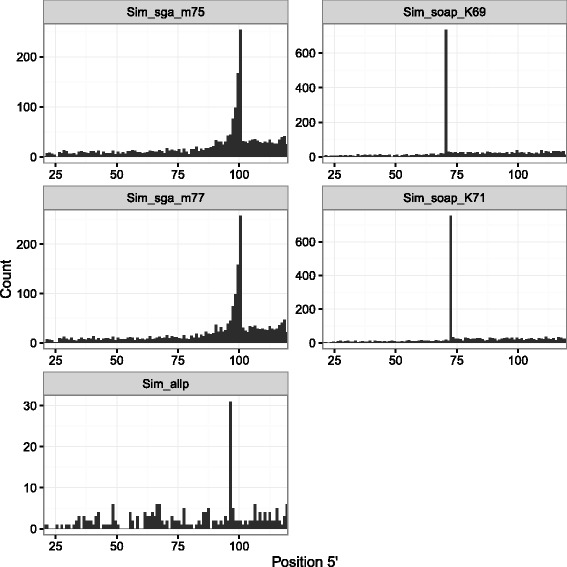
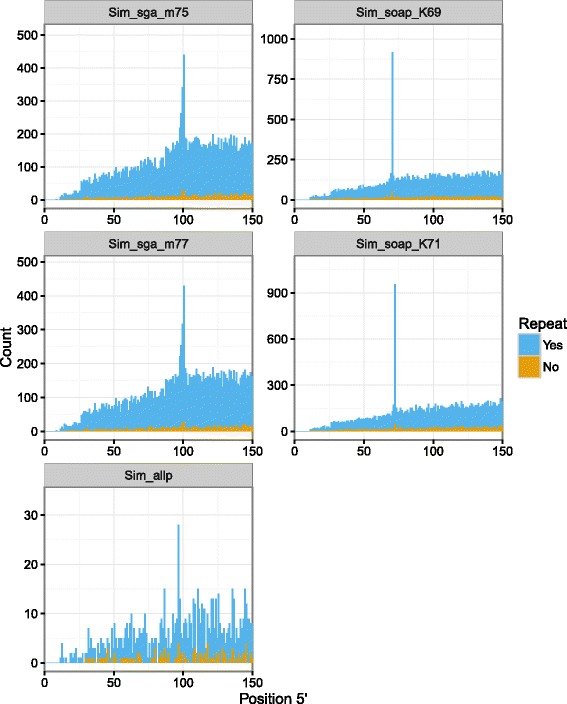
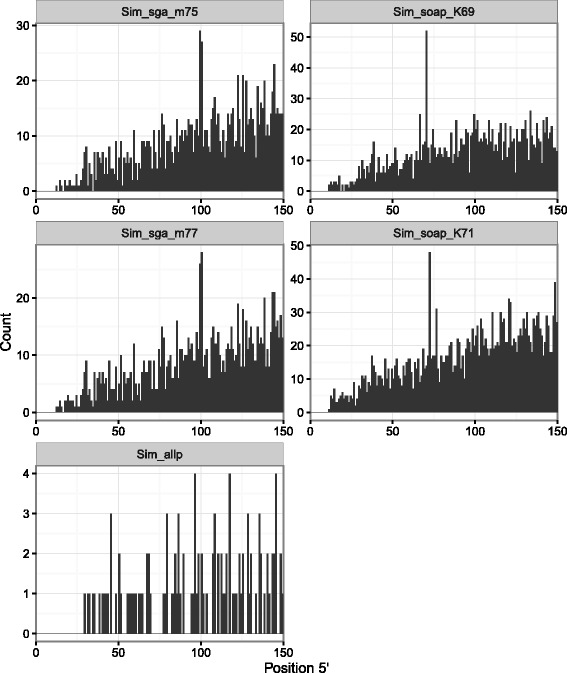
Results: In this article, we report a positional bias present in variant calling performed against draft reference assemblies constructed from de Bruijn or string overlap graphs. We assessed how frequently variants appeared at each position counted from ends of a contig or scaffold sequence, and discovered unexpectedly high number of variants at the positions related to the length of either k-mers or reads used for the assembly. We detected the bias in both publicly available draft assemblies from Assemblathon 2 competition as well as in the assemblies we generated from our simulated short-read data. Simulations confirmed that the bias causing variants are predominantly false positives induced by reads from spatially distant repeated sequences. The bias is particularly strong in contig assemblies. Scaffolding does not eliminate the bias but tends to mitigate it because of the changes in variants' relative positions and alterations in read alignments. The bias can be effectively reduced by filtering out the variants that reside in repetitive elements.
Conclusions: Draft genome sequences generated by several popular assemblers appear to be susceptible to the positional bias potentially affecting many resequencing projects in non-model species. The bias is inherent to the assembly algorithms and arises from their particular handling of repeated sequences. It is recommended to reduce the bias by filtering especially if higher-quality genome assembly cannot be achieved. Our findings can help other researchers to improve the quality of their variant data sets and reduce artefactual findings in downstream analyses.
Keywords: Draft reference genome; Polymorphisms; Positional bias; Repetitive elements; Reseqencing; SNPs; Variants.
Figures
References
-
- Chia JM, Song C, Bradbury PJ, Costich D, de Leon N, Doebley J, et al. Maize HapMap2 identifies extant variation from a genome in flux. Nature Genetics. 2012; 44(7):803–807. Available from: http://dx.doi.org/10.1038/ng.2313. - DOI - PubMed
-
- Stanton-Geddes J, Paape T, Epstein B, Briskine R, Yoder J, Mudge J, et al. Candidate genes and genetic architecture of symbiotic and agronomic traits revealed by whole-genome, sequence-based association genetics in Medicago truncatula. PLoS ONE. 2013; 8(5):Available from: http://dx.doi.org/10.1371/journal.pone.0065688. - DOI - PMC - PubMed
-
- Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nature Genetics. 2015; 47(5):435–444. Available from: http://dx.doi.org/10.1038/ng.3247. - DOI - PubMed
-
- Rondeau EB, Minkley DR, Leong JS, Messmer AM, Jantzen JR, von Schalburg KR, et al. The genome and linkage map of the northern pike (Esox lucius): conserved synteny revealed between the salmonid sister group and the Neoteleostei. PLoS ONE. 2014; Jul;9(7):e102089. Available from: http://dx.doi.org/10.1371/journal.pone.0102089. - DOI - PMC - PubMed
-
- Nowak M, Russo G, Schlapbach R, Huu C, Lenhard M, Conti E. The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biology. 2015; 16(1):12. Available from: http://genomebiology.com/2015/16/1/12. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
