

Cardiac Function Remains Impaired Despite Reversible Cardiac Remodeling after Acute Experimental Viral Myocarditis
- PMID: 28352641
- PMCID: PMC5352897
- DOI: 10.1155/2017/6590609
Cardiac Function Remains Impaired Despite Reversible Cardiac Remodeling after Acute Experimental Viral Myocarditis
Abstract
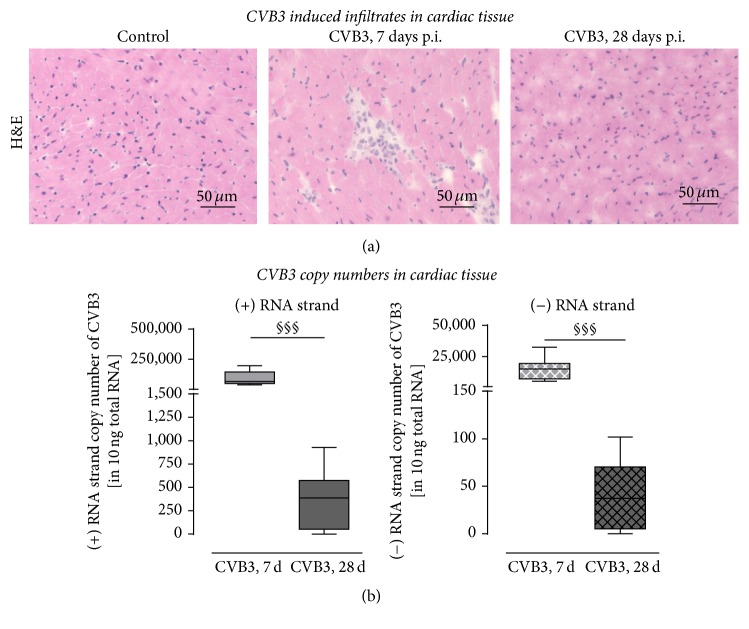
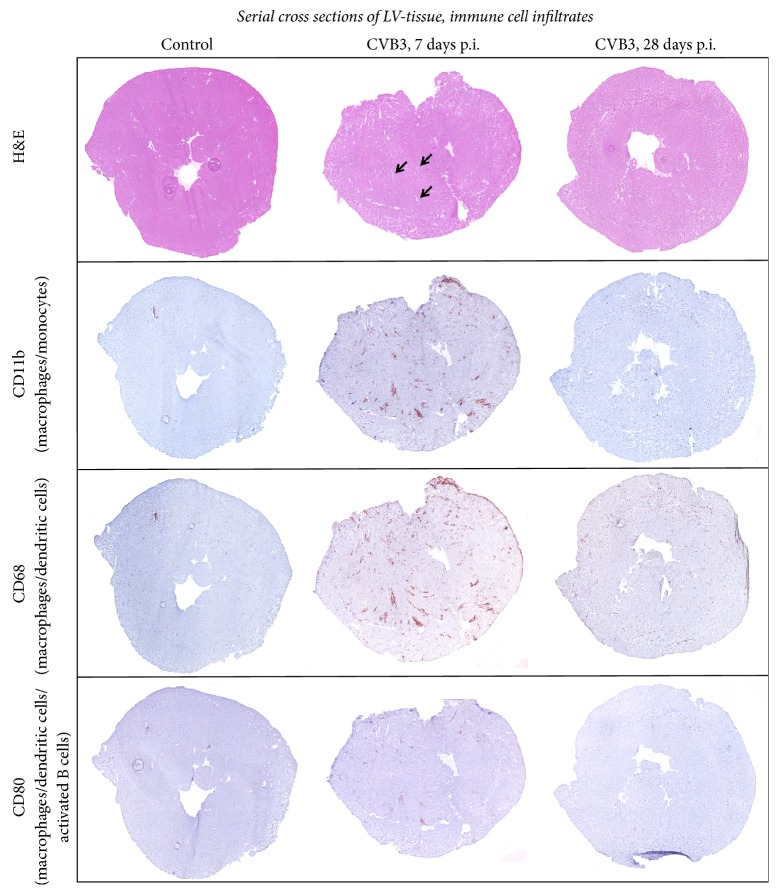
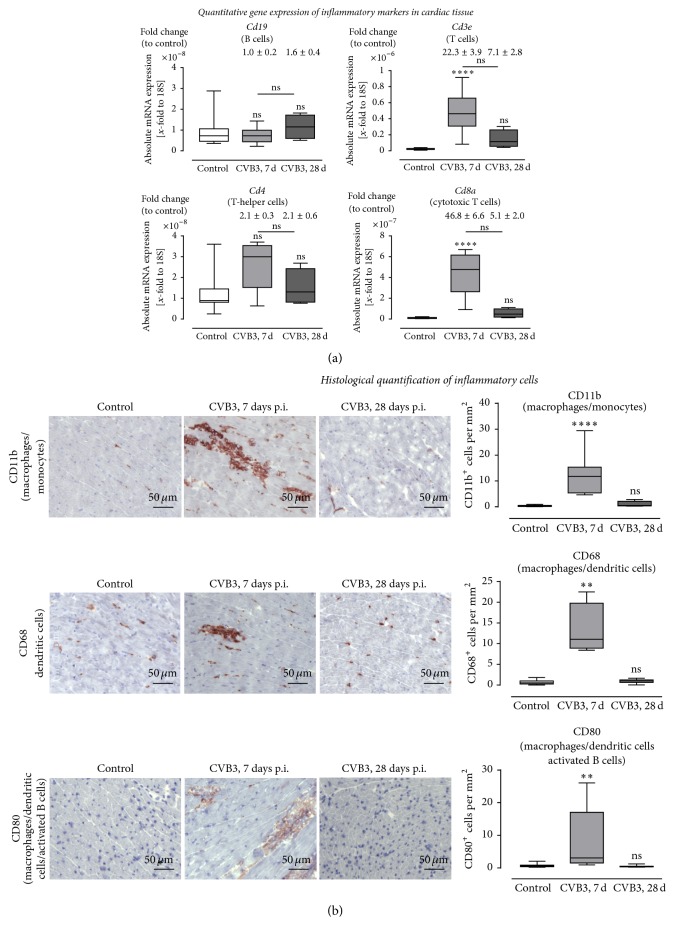
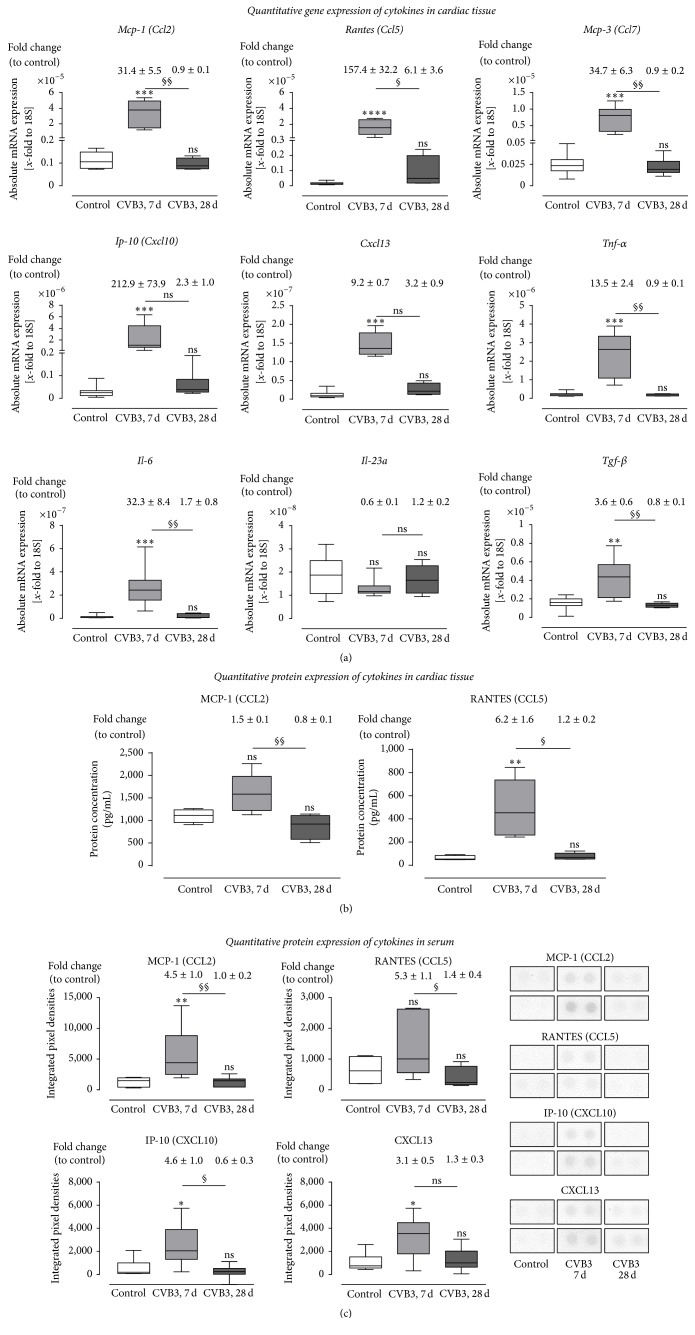
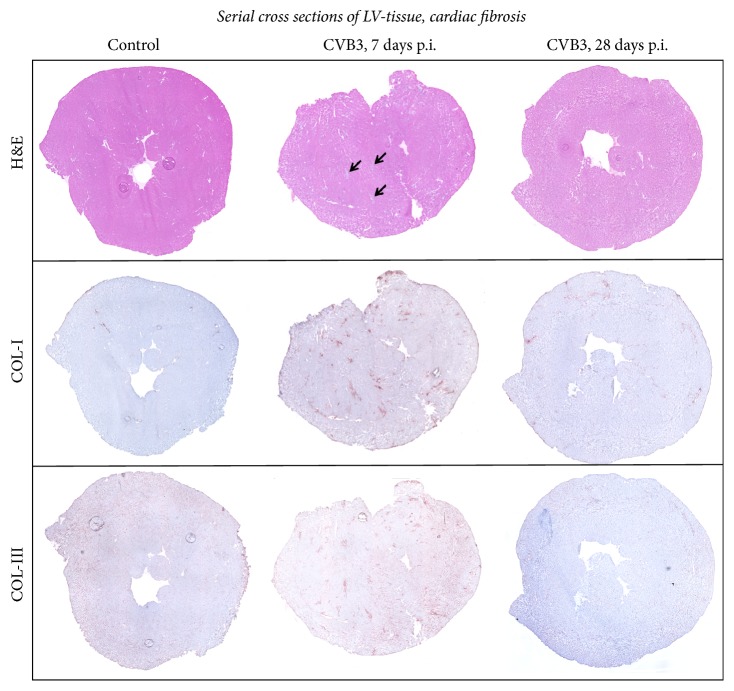
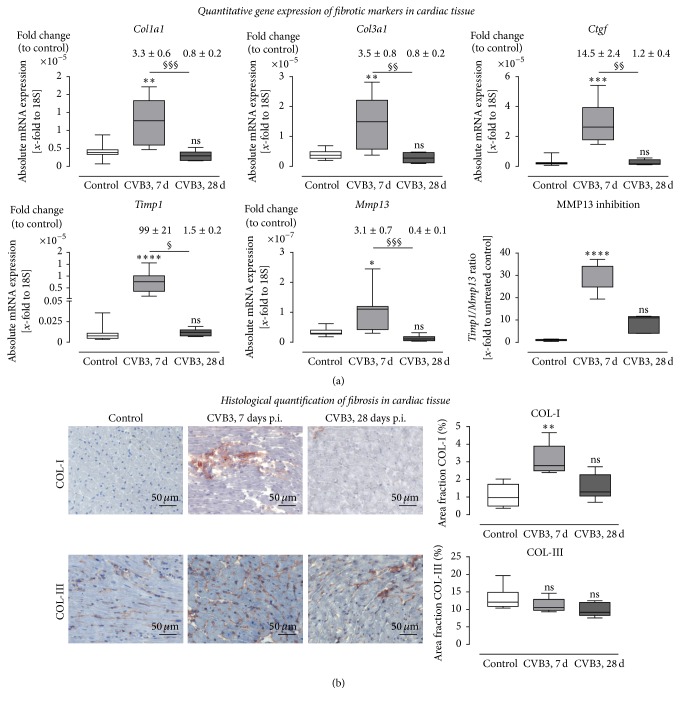
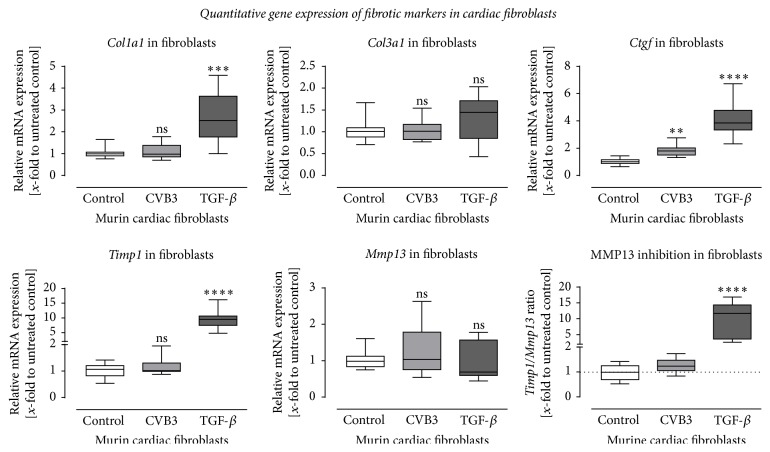
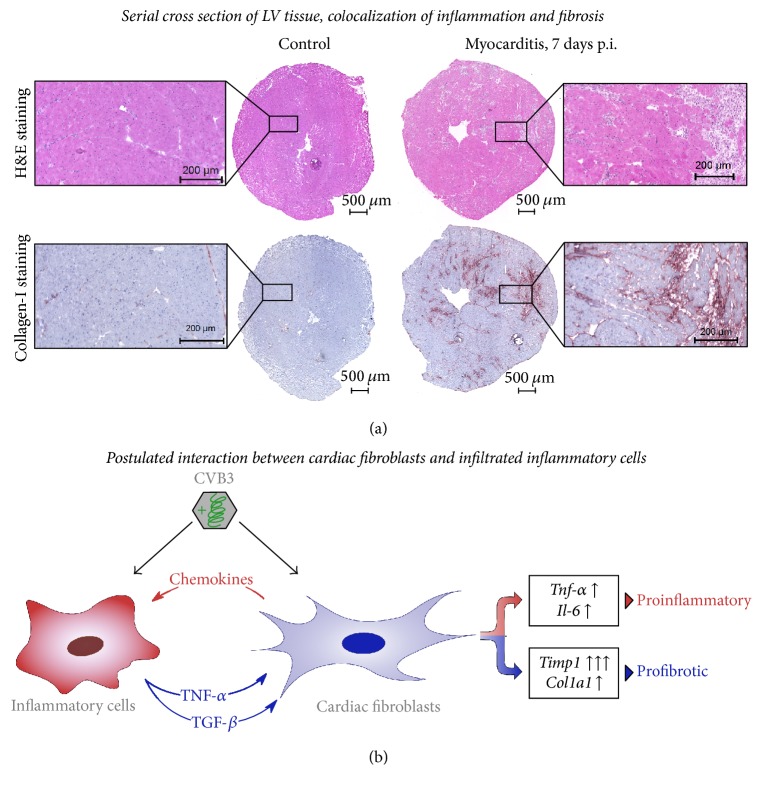
Background. Infection with Coxsackievirus B3 induces myocarditis. We aimed to compare the acute and chronic phases of viral myocarditis to identify the immediate effects of cardiac inflammation as well as the long-term effects after resolved inflammation on cardiac fibrosis and consequently on cardiac function. Material and Methods. We infected C57BL/6J mice with Coxsackievirus B3 and determined the hemodynamic function 7 as well as 28 days after infection. Subsequently, we analyzed viral burden and viral replication in the cardiac tissue as well as the expression of cytokines and matrix proteins. Furthermore, cardiac fibroblasts were infected with virus to investigate if viral infection alone induces profibrotic signaling. Results. Severe cardiac inflammation was determined and cardiac fibrosis was consistently colocalized with inflammation during the acute phase of myocarditis. Declined cardiac inflammation but no significantly improved hemodynamic function was observed 28 days after infection. Interestingly, cardiac fibrosis declined to basal levels as well. Both cardiac inflammation and fibrosis were reversible, whereas the hemodynamic function remains impaired after healed viral myocarditis in C57BL/6J mice.
Conflict of interest statement
The authors declare that there is no conflict of interests regarding the publication of this paper.
Figures
References
-
- Caforio A. L. P., Pankuweit S., Arbustini E., et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European Heart Journal. 2013;34(33):2636–2648. doi: 10.1093/eurheartj/eht210. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
