

Ephemeral Protein Binding to DNA Shapes Stable Nuclear Bodies and Chromatin Domains
- PMID: 28355537
- PMCID: PMC5375003
- DOI: 10.1016/j.bpj.2017.01.025
Ephemeral Protein Binding to DNA Shapes Stable Nuclear Bodies and Chromatin Domains
Abstract
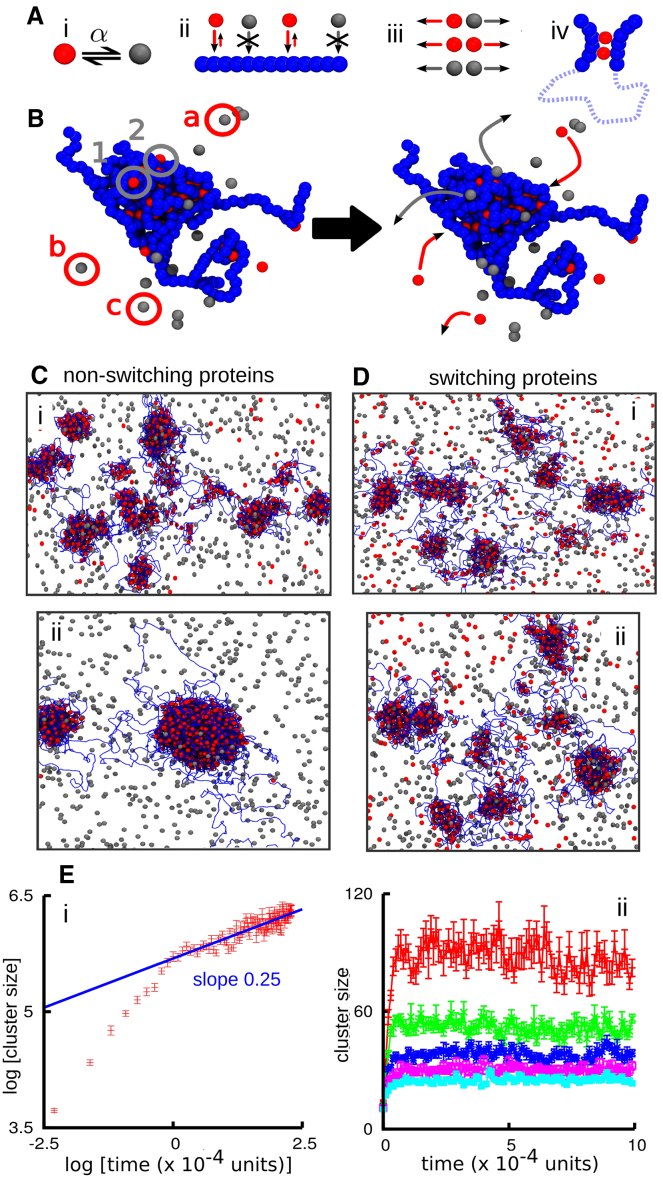
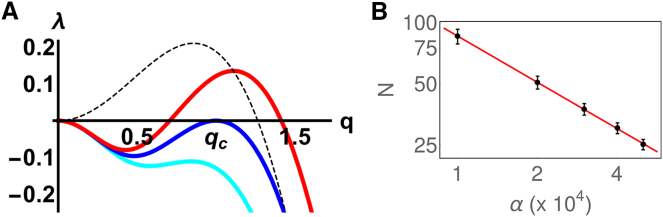
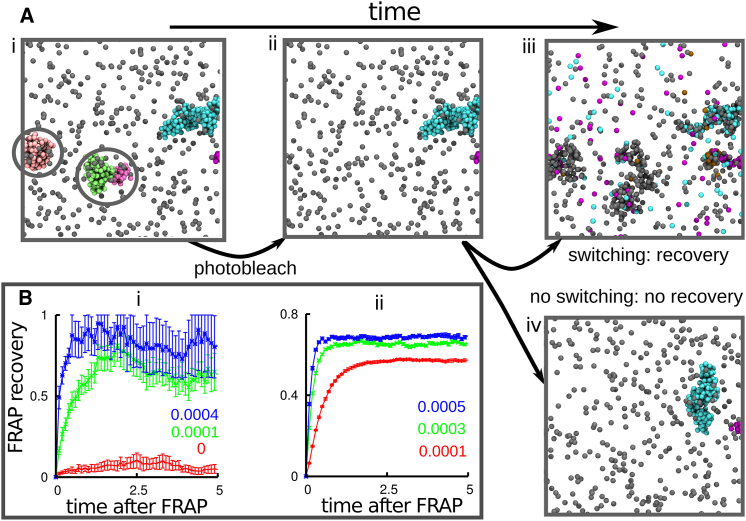
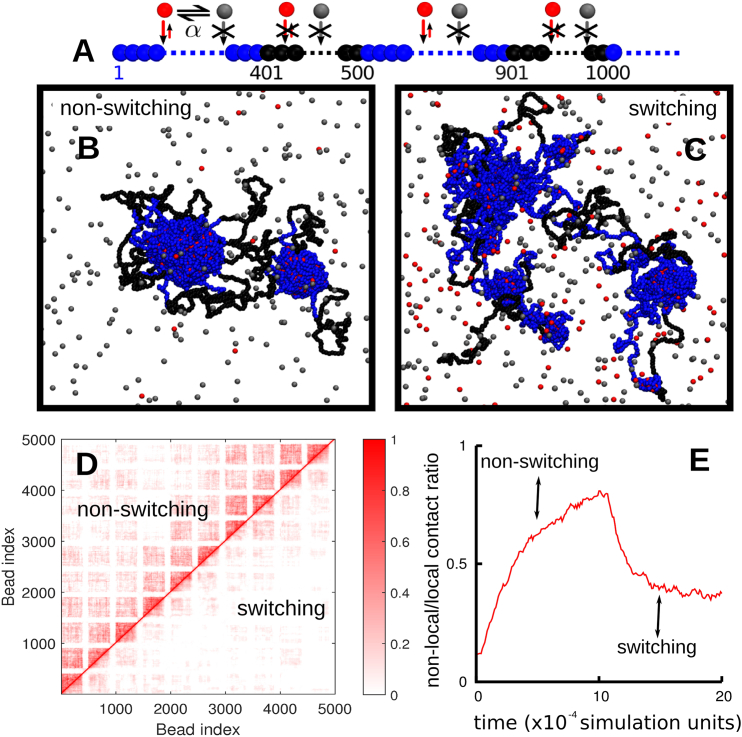
Fluorescence microscopy reveals that the contents of many (membrane-free) nuclear bodies exchange rapidly with the soluble pool while the underlying structure persists; such observations await a satisfactory biophysical explanation. To shed light on this, we perform large-scale Brownian dynamics simulations of a chromatin fiber interacting with an ensemble of (multivalent) DNA-binding proteins able to switch between an "on" (binding) and an "off" (nonbinding) state. This system provides a model for any DNA-binding protein that can be posttranslationally modified to change its affinity for DNA (e.g., through phosphorylation). Protein switching is a nonequilibrium process, and it leads to the formation of clusters of self-limiting size, where individual proteins in a cluster exchange with the soluble pool with kinetics similar to those seen in photobleaching experiments. This behavior contrasts sharply with that exhibited by nonswitching proteins, which are permanently in the on-state; when these bind to DNA nonspecifically, they form clusters that grow indefinitely in size. To explain these findings, we propose a mean-field theory from which we obtain a scaling relation between the typical cluster size and the protein switching rate. Protein switching also reshapes intrachromatin contacts to give networks resembling those seen in topologically associating domains, as switching markedly favors local (short-range) contacts over distant ones. Our results point to posttranslational modification of chromatin-bridging proteins as a generic mechanism driving the self-assembly of highly dynamic, nonequilibrium, protein clusters with the properties of nuclear bodies.
Copyright © 2017 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Calladine C.R., Drew H.R., Travers A.A. Ed. 3. Elsevier; London, UK: 2004. Understanding DNA.
-
- Alberts B. Garland Science; New York: 2002. Molecular Biology of the Cell.
-
- Cook P.R. Wiley-Liss; New York: 2001. Principles of Nuclear Structure and Function.
-
- Dame R.T., Noom M.C., Wuite G.J. Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature. 2006;444:387–390. - PubMed
-
- Simonis M., Klous P., de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat. Genet. 2006;38:1348–1354. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
