

A Sequentially Priming Phosphorylation Cascade Activates the Gliomagenic Transcription Factor Olig2
- PMID: 28355568
- PMCID: PMC5394178
- DOI: 10.1016/j.celrep.2017.03.003
A Sequentially Priming Phosphorylation Cascade Activates the Gliomagenic Transcription Factor Olig2
Abstract
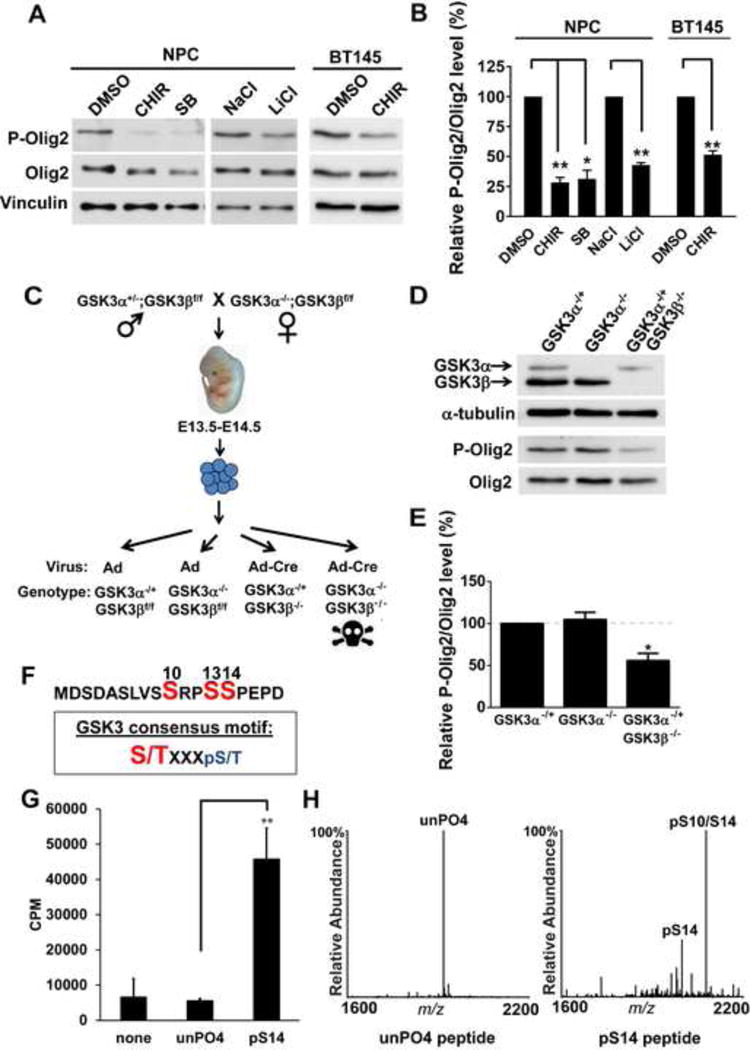
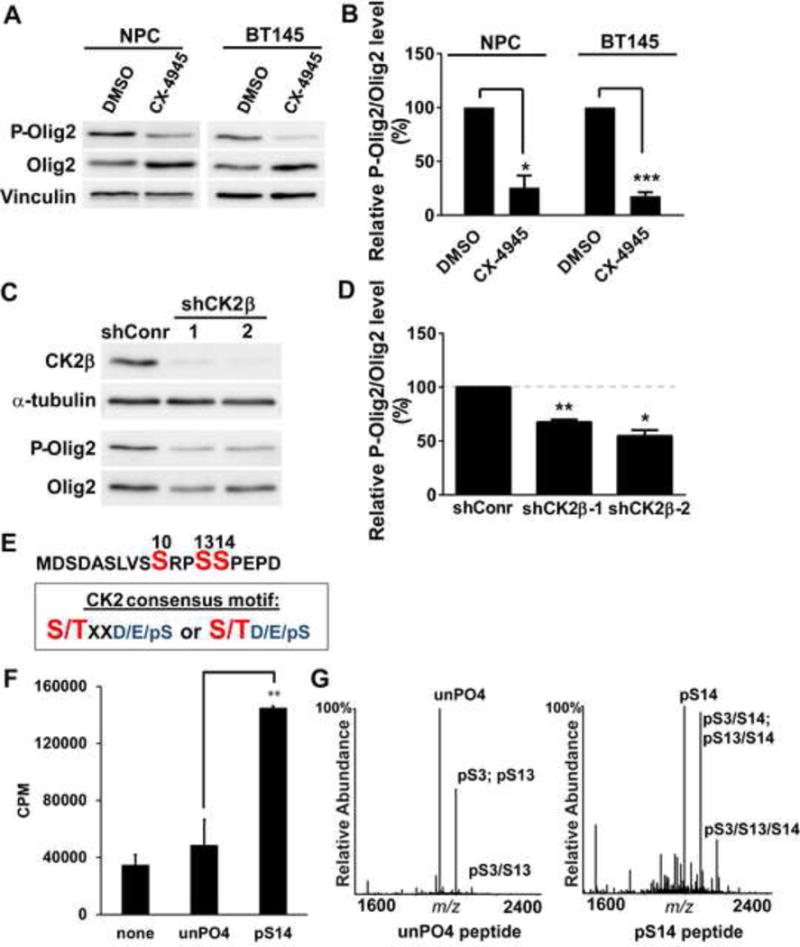
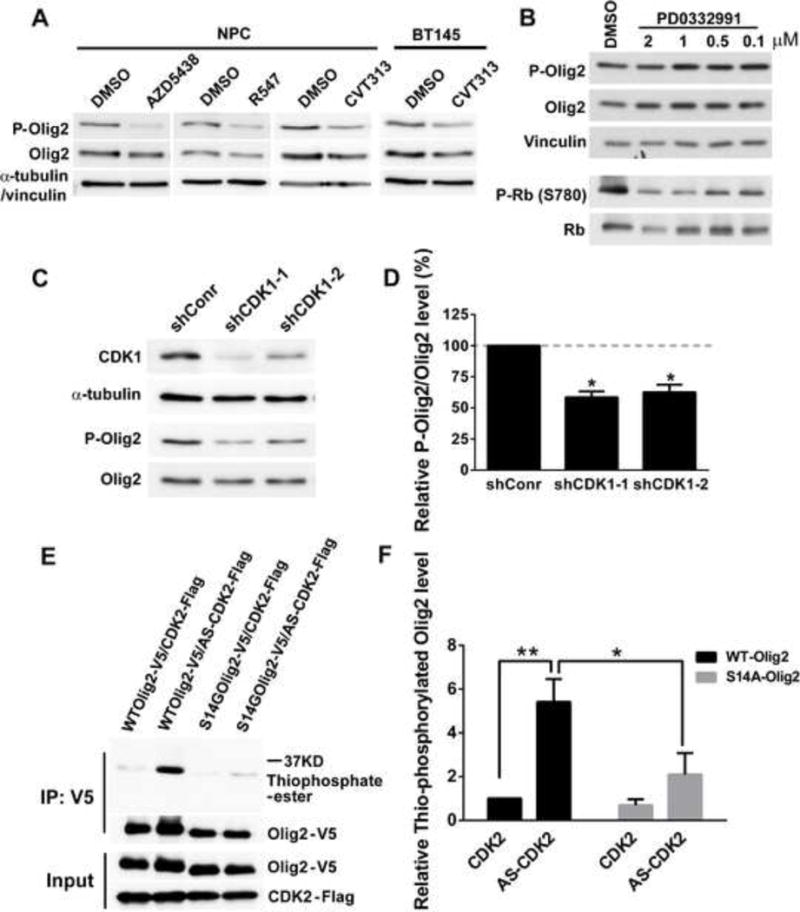
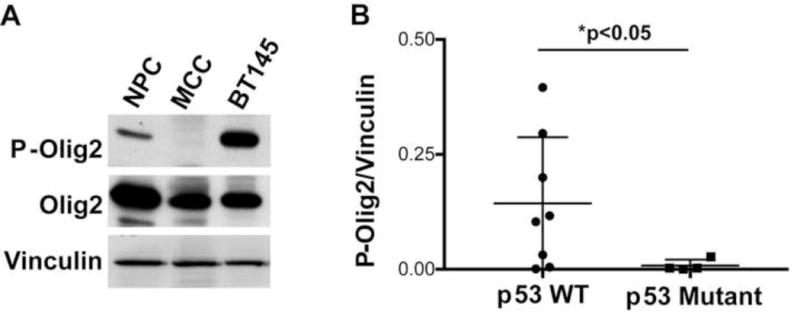
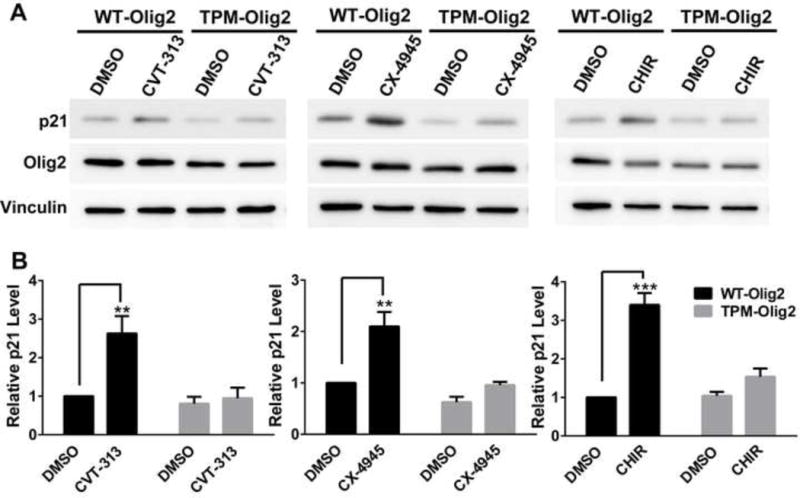
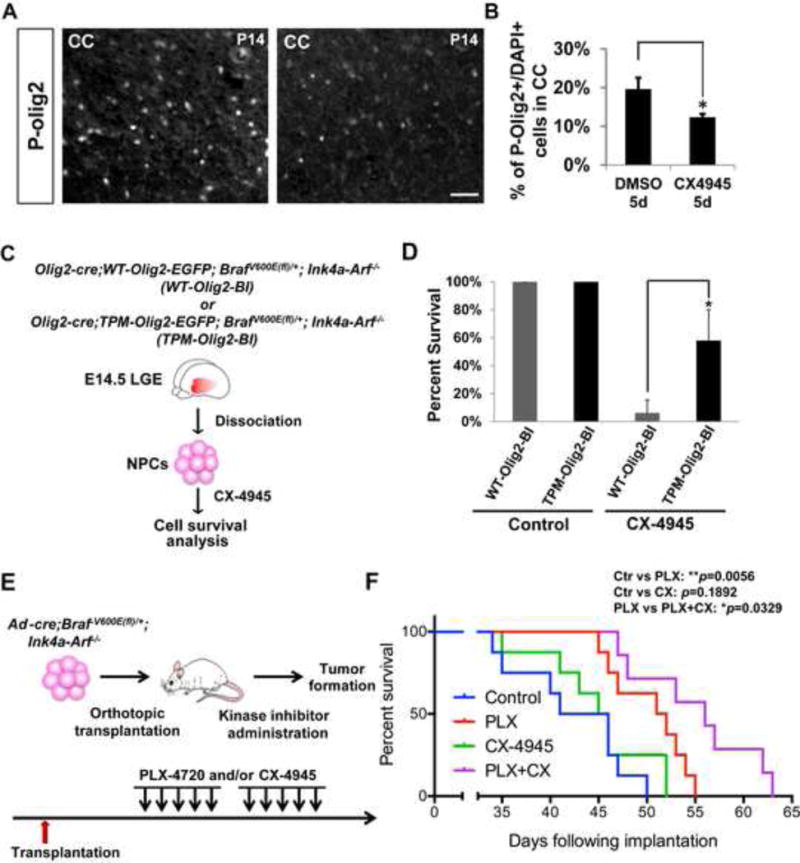
During development of the vertebrate CNS, the basic helix-loop-helix (bHLH) transcription factor Olig2 sustains replication competence of progenitor cells that give rise to neurons and oligodendrocytes. A pathological counterpart of this developmental function is seen in human glioma, wherein Olig2 is required for maintenance of stem-like cells that drive tumor growth. The mitogenic/gliomagenic functions of Olig2 are regulated by phosphorylation of a triple serine motif (S10, S13, and S14) in the amino terminus. Here, we identify a set of three serine/threonine protein kinases (glycogen synthase kinase 3α/β [GSK3α/β], casein kinase 2 [CK2], and cyclin-dependent kinases 1/2 [CDK1/2]) that are, collectively, both necessary and sufficient to phosphorylate the triple serine motif. We show that phosphorylation of the motif itself serves as a template to prime phosphorylation of additional serines and creates a highly charged "acid blob" in the amino terminus of Olig2. Finally, we show that small molecule inhibitors of this forward-feeding phosphorylation cascade have potential as glioma therapeutics.
Keywords: CDK; CK2; GSK3; NPCs; Olig2; casein kinase 2; cyclin-dependent kinase; glioma; glycogen synthase kinase 3; neural progenitor cells; phosphorylation; protein kinase.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. - PubMed
-
- Bouvier C, Bartoli C, Aguirre-Cruz L, Virard I, Colin C, Fernandez C, Gouvernet J, Figarella-Branger D. Shared oligodendrocyte lineage gene expression in gliomas and oligodendrocyte progenitor cells. J Neurosurg. 2003;99:344–350. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
