

Severity of Demyelinating and Axonal Neuropathy Mouse Models Is Modified by Genes Affecting Structure and Function of Peripheral Nodes
- PMID: 28355569
- PMCID: PMC5415377
- DOI: 10.1016/j.celrep.2017.03.009
Severity of Demyelinating and Axonal Neuropathy Mouse Models Is Modified by Genes Affecting Structure and Function of Peripheral Nodes
Abstract
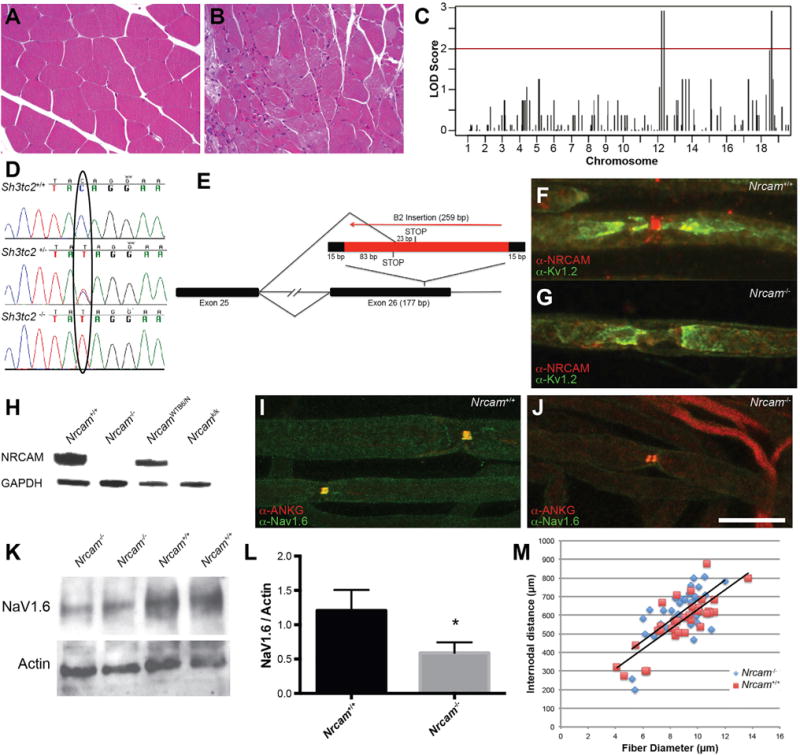
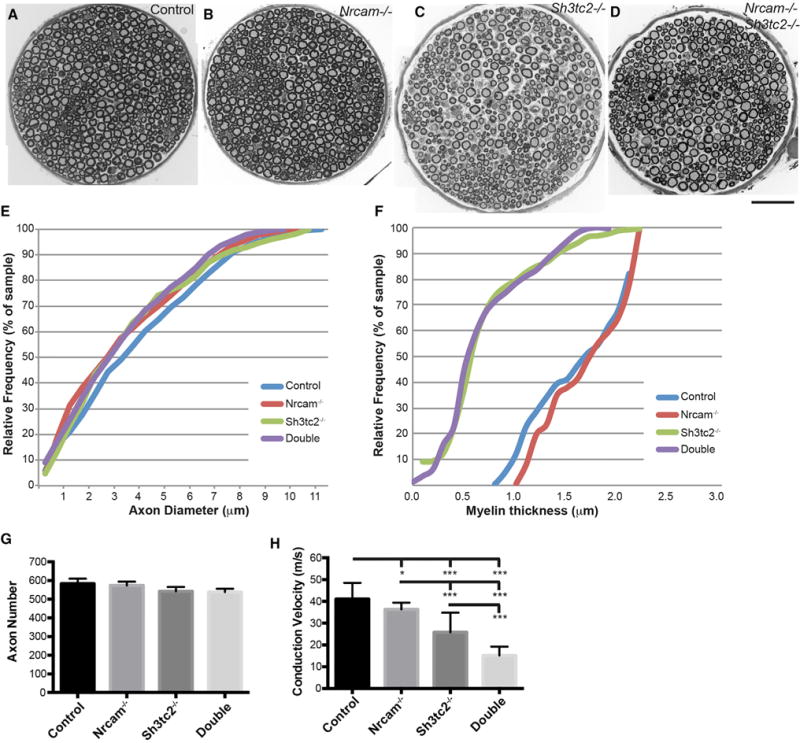
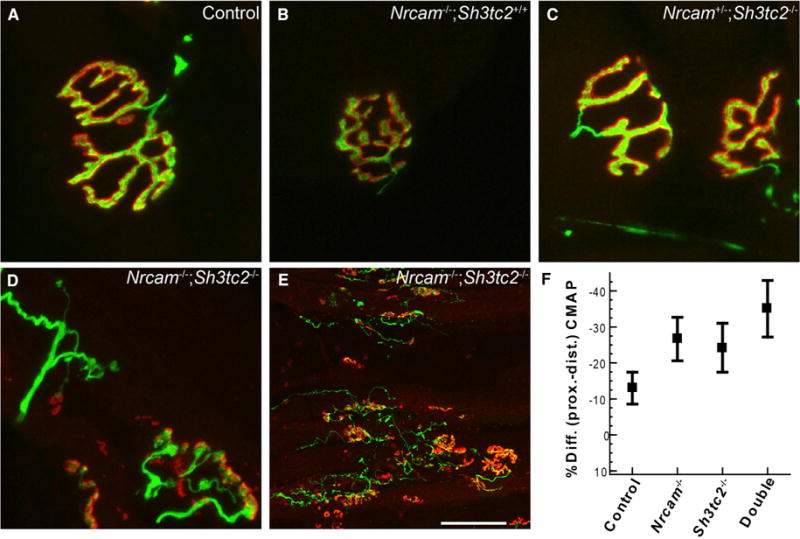
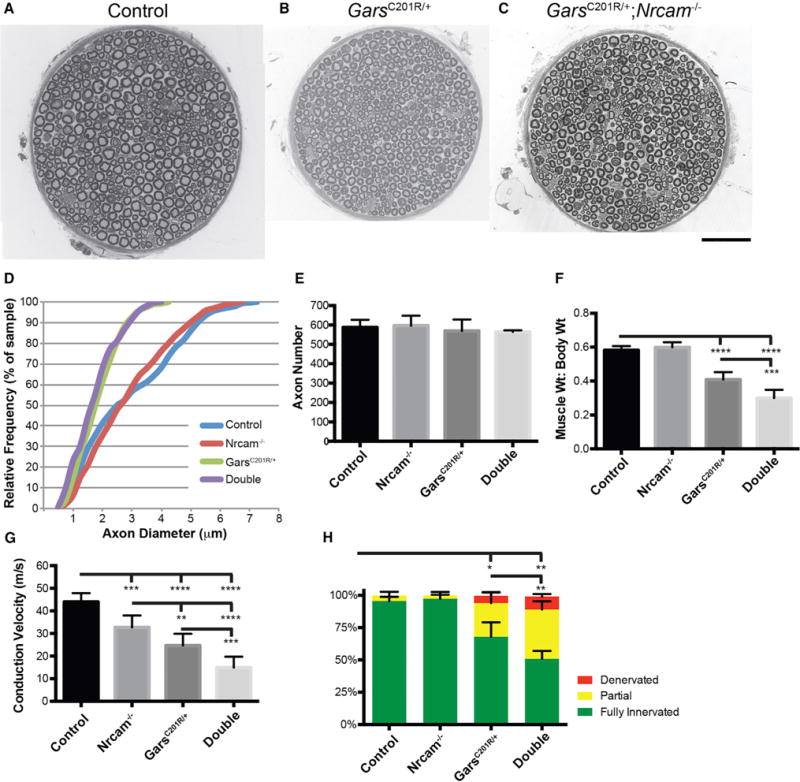
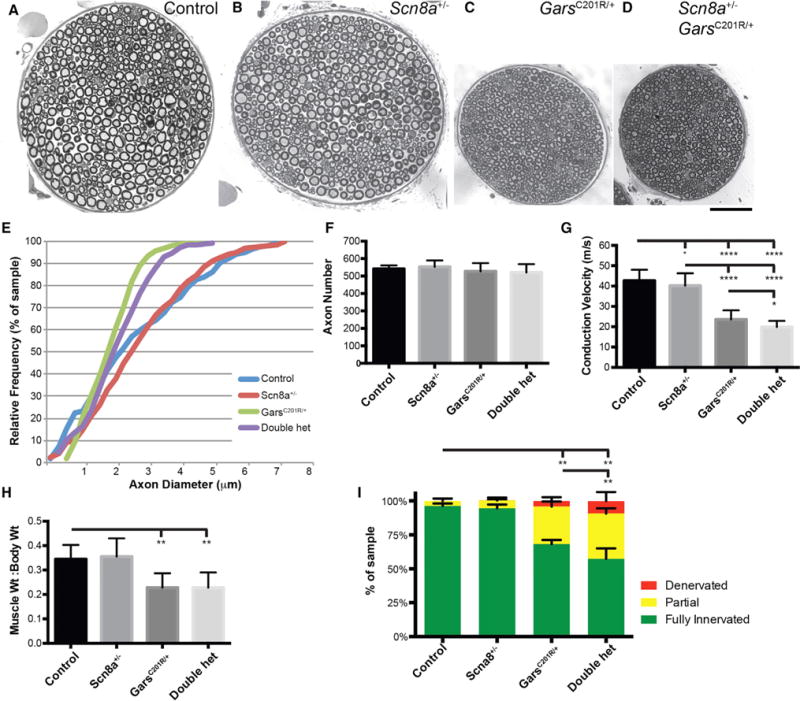
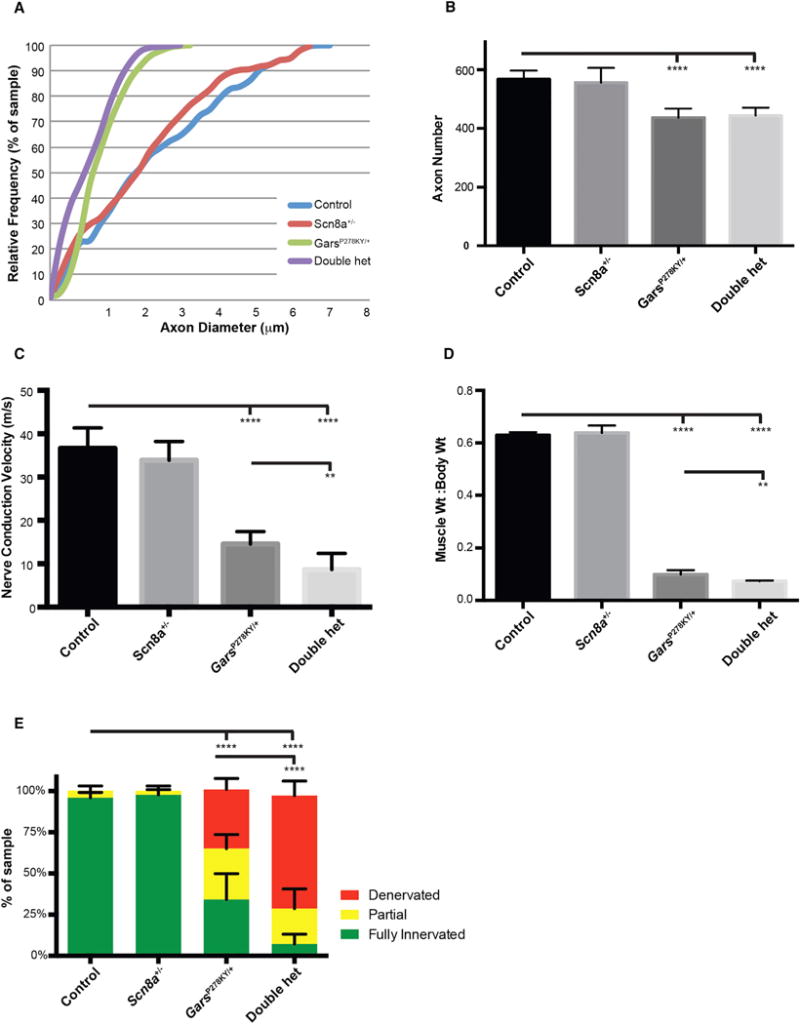
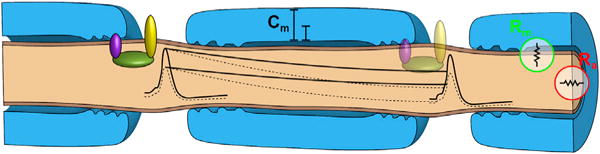
Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heterogeneous group of inherited polyneuropathies. Mutations in 80 genetic loci can cause forms of CMT, resulting in demyelination and axonal dysfunction. The clinical presentation, including sensory deficits, distal muscle weakness, and atrophy, can vary greatly in severity and progression. Here, we used mouse models of CMT to demonstrate genetic interactions that result in a more severe neuropathy phenotype. The cell adhesion molecule Nrcam and the Na+ channel Scn8a (NaV1.6) are important components of nodes. Homozygous Nrcam and heterozygous Scn8a mutations synergized with both an Sh3tc2 mutation, modeling recessive demyelinating Charcot-Marie-Tooth type 4C, and mutations in Gars, modeling dominant axonal Charcot-Marie-Tooth type 2D. We conclude that genetic variants perturbing the structure and function of nodes interact with mutations affecting the cable properties of axons by thinning myelin or reducing axon diameter. Therefore, genes integral to peripheral nodes are candidate modifiers of peripheral neuropathy.
Keywords: Charcot-Marie-Tooth disease; axons; degeneration; genetic modifiers; hereditary sensory; length constant; motor neuropathy.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Achilli F, Bros-Facer V, Williams HP, Banks GT, AlQatari M, Chia R, Tucci V, Groves M, Nickols CD, Seburn KL, et al. An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis Model Mech. 2009;2:359–373. - PMC - PubMed
-
- Arnaud E, Zenker J, de Preux Charles AS, Stendel C, Roos A, Médard JJ, Tricaud N, Kleine H, Luscher B, Weis J, et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci USA. 2009;106:17528–17533. - PMC - PubMed
-
- Bonora E, Lamb JA, Barnby G, Sykes N, Moberly T, Beyer KS, Klauck SM, Poustka F, Bacchelli E, Blasi F, et al. Mutation screening and association analysis of six candidate genes for autism on chromosome 7q. Eur J Hum Genet. 2005;13:198–207. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
