

Cytokine receptor signaling is required for the survival of ALK- anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations
- PMID: 28356514
- PMCID: PMC5393253
- DOI: 10.1073/pnas.1700682114
Cytokine receptor signaling is required for the survival of ALK- anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations
Abstract
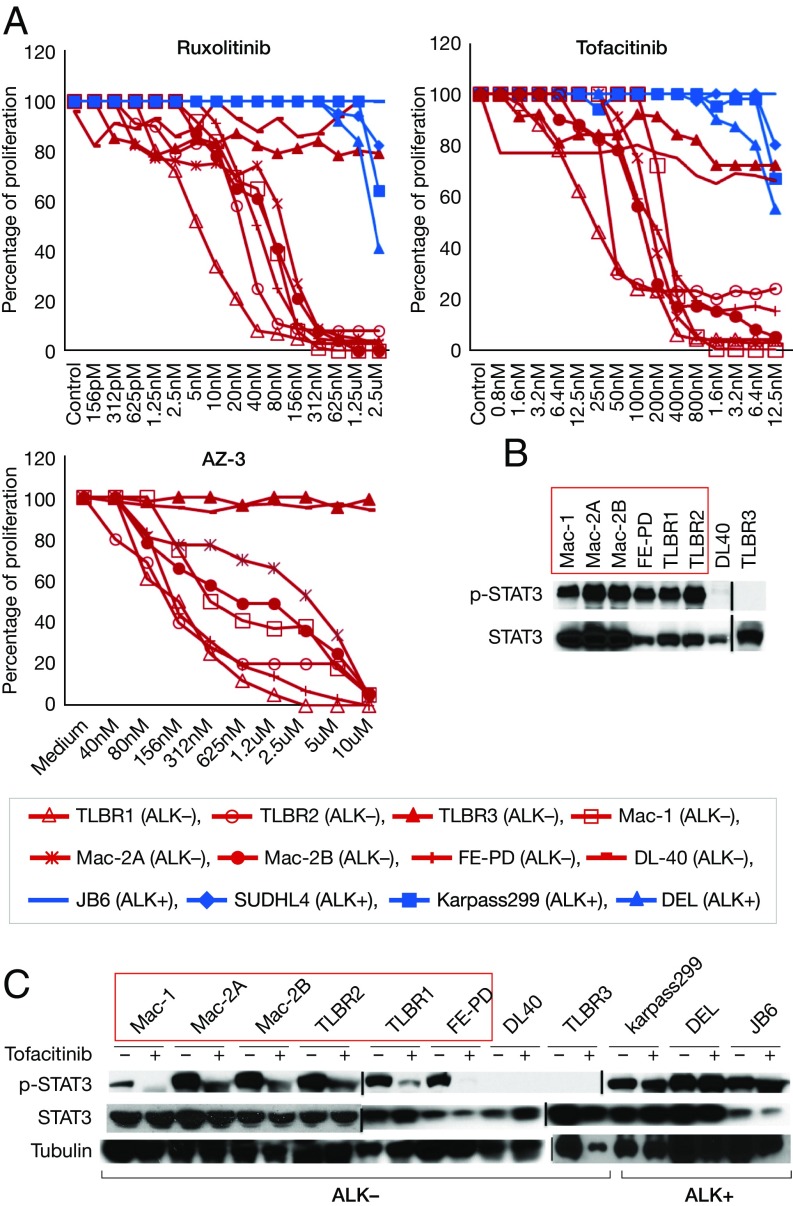
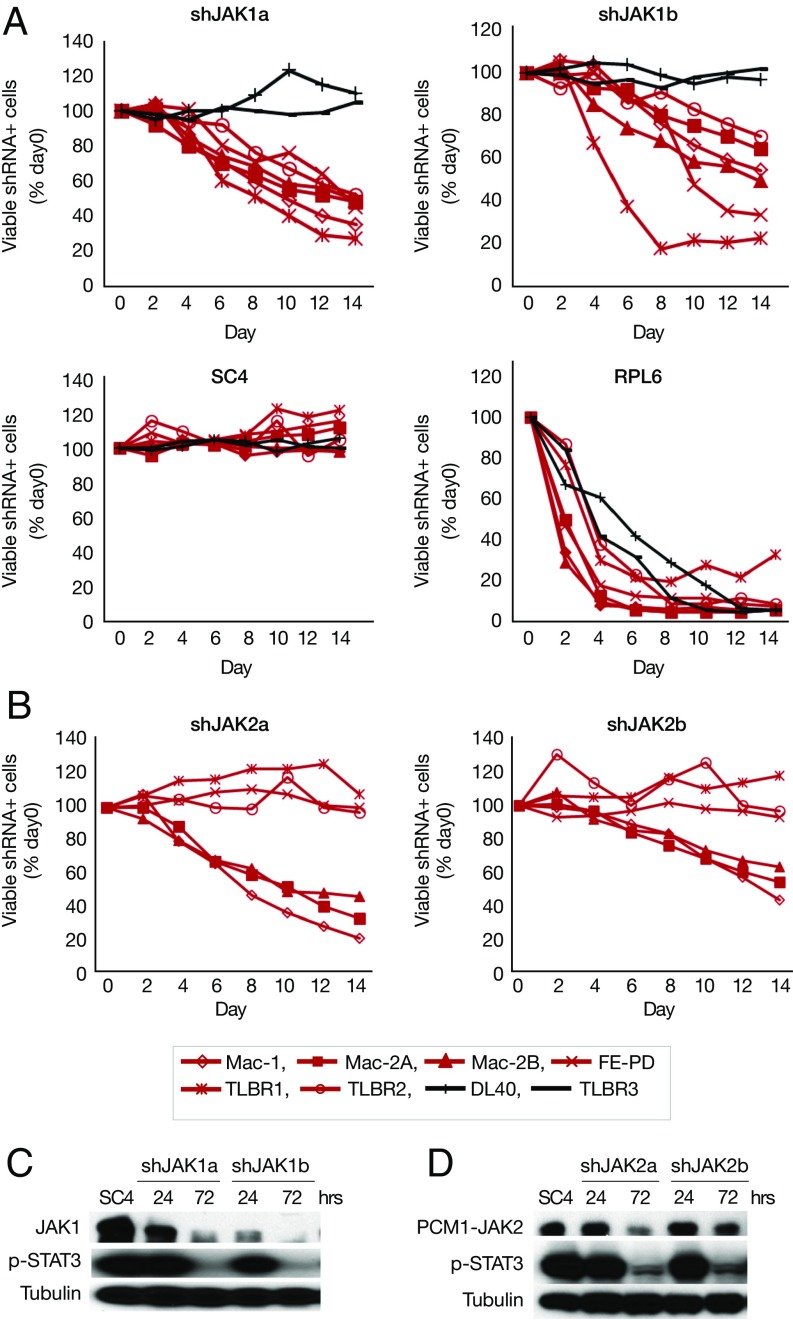
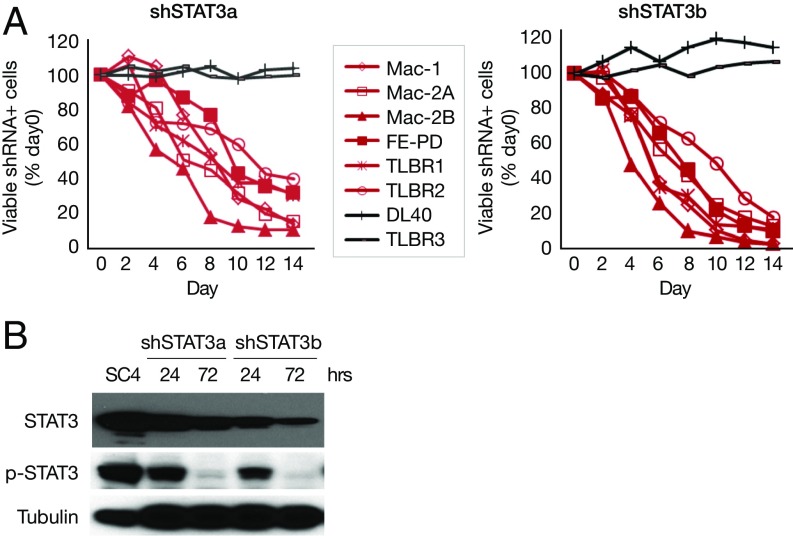
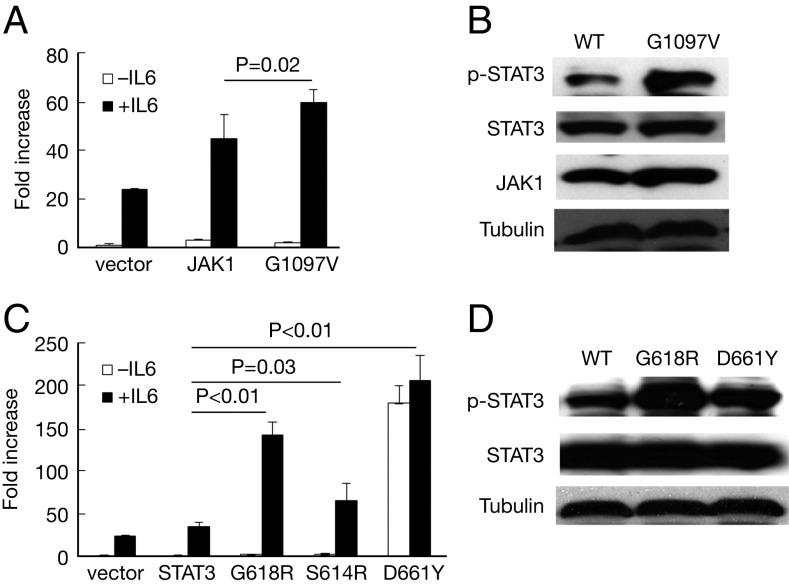
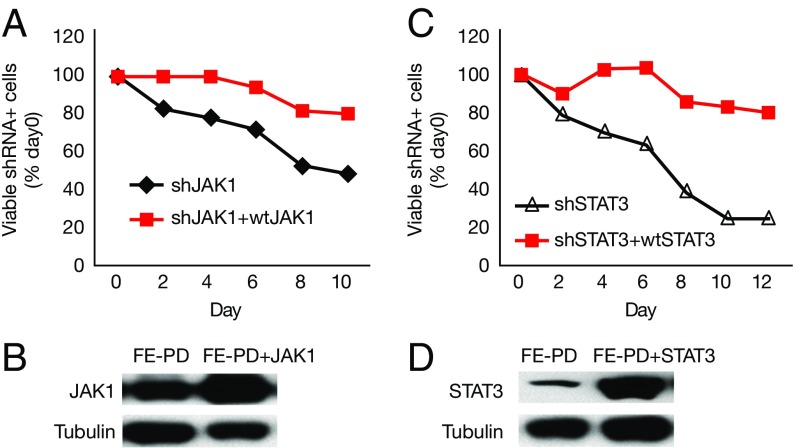
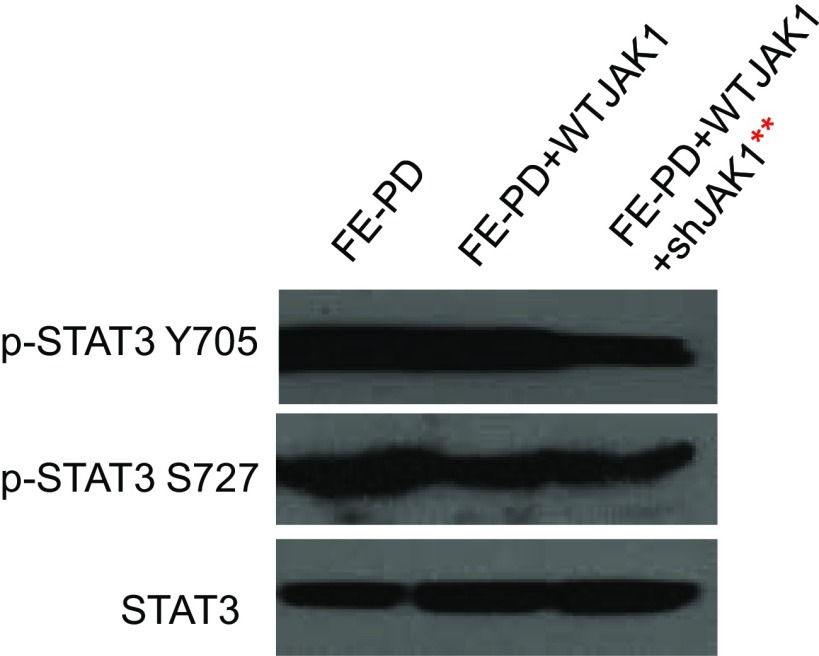
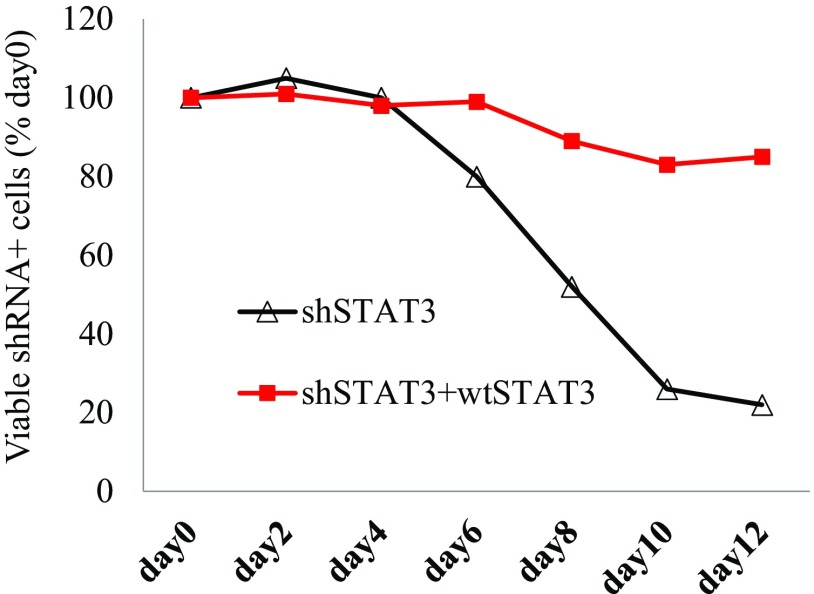
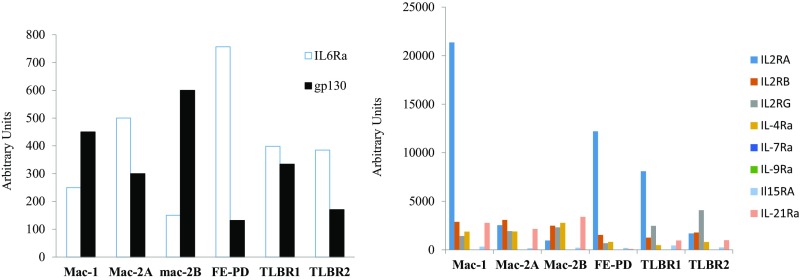
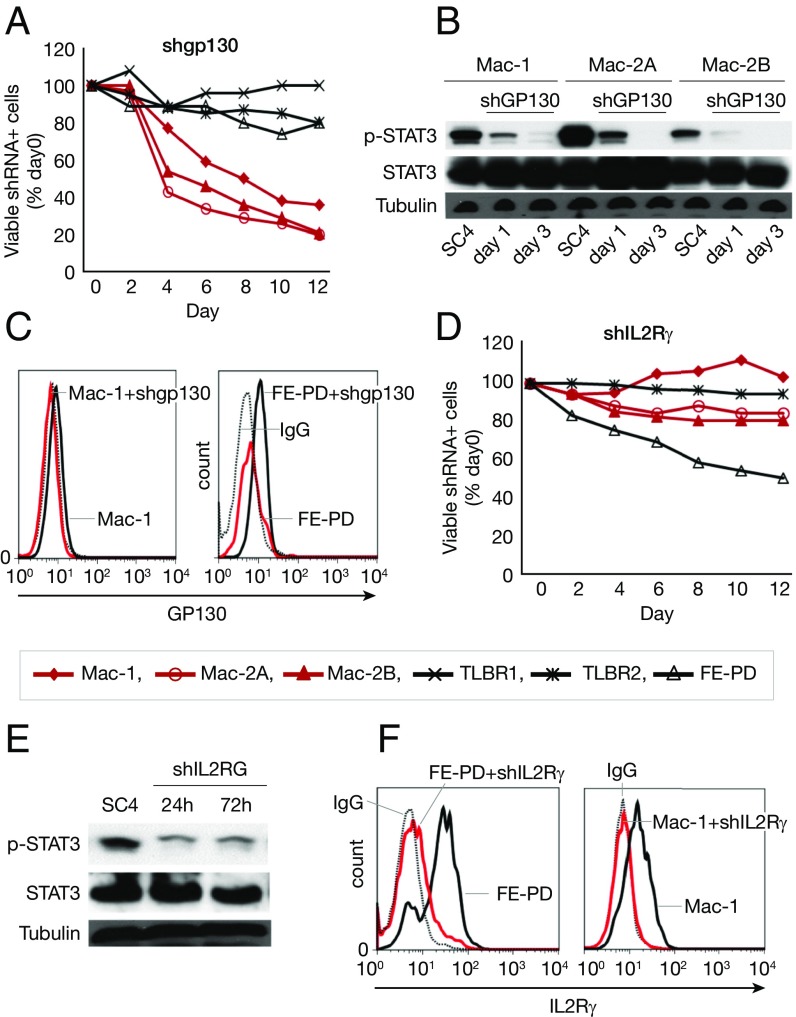
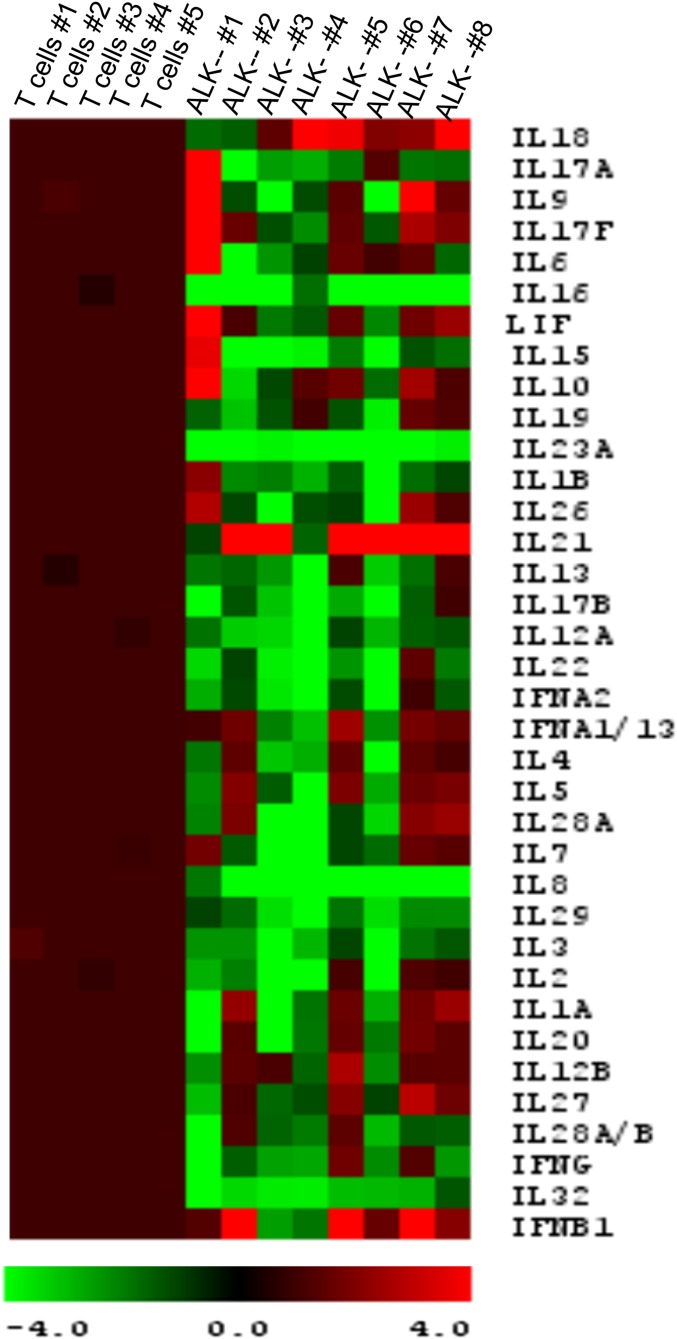
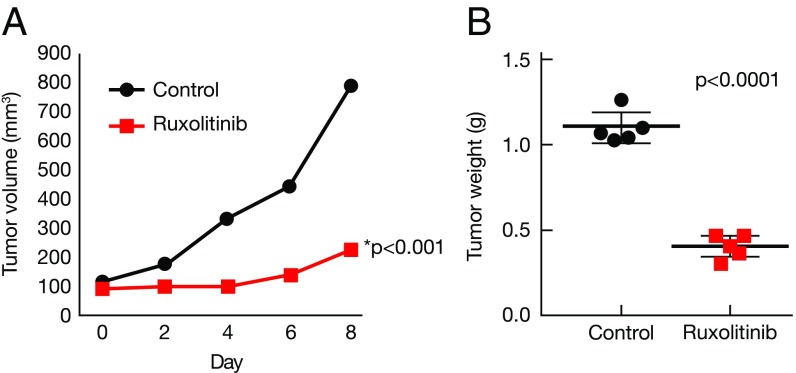
Activating Janus kinase (JAK) and signal transducer and activator of transcription (STAT) mutations have been discovered in many T-cell malignancies, including anaplastic lymphoma kinase (ALK)- anaplastic large cell lymphomas (ALCLs). However, such mutations occur in a minority of patients. To investigate the clinical application of targeting JAK for ALK- ALCL, we treated ALK- cell lines of various histological origins with JAK inhibitors. Interestingly, most exogenous cytokine-independent cell lines responded to JAK inhibition regardless of JAK mutation status. JAK inhibitor sensitivity correlated with the STAT3 phosphorylation status of tumor cells. Using retroviral shRNA knockdown, we have demonstrated that these JAK inhibitor-sensitive cells are dependent on both JAK1 and STAT3 for survival. JAK1 and STAT3 gain-of-function mutations were found in some, but not all, JAK inhibitor-sensitive cells. Moreover, the mutations alone cannot explain the JAK1/STAT3 dependency, given that wild-type JAK1 or STAT3 was sufficient to promote cell survival in the cells that had either JAK1or STAT3 mutations. To investigate whether other mechanisms were involved, we knocked down upstream receptors GP130 or IL-2Rγ. Knockdown of GP130 or IL-2Rγ induced cell death in selected JAK inhibitor-sensitive cells. High expression levels of cytokines, including IL-6, were demonstrated in cell lines as well as in primary ALK- ALCL tumors. Finally, ruxolitinib, a JAK1/2 inhibitor, was effective in vivo in a xenograft ALK- ALCL model. Our data suggest that cytokine receptor signaling is required for tumor cell survival in diverse forms of ALK- ALCL, even in the presence of JAK1/STAT3 mutations. Therefore, JAK inhibitor therapy might benefit patients with ALK- ALCL who are phosphorylated STAT3<sup/>.
Keywords: ALK− ALCL; JAK; STAT; cytokine receptor; mutations.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Kempf W. CD30+ lymphoproliferative disorders: Histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(Suppl 1):58–70. - PubMed
-
- Morris SW, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284. - PubMed
-
- Chiarle R, et al. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat Med. 2008;14(6):676–680. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
