

Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark
- PMID: 28357410
- PMCID: PMC5362145
- DOI: 10.1212/NXG.0000000000000140
Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark
Abstract
Objective: Congenital myopathy as a nosologic entity has long been recognized, but knowledge of overall and subtype prevalence and phenotype-genotype relationship is scarce, especially in the adult population.
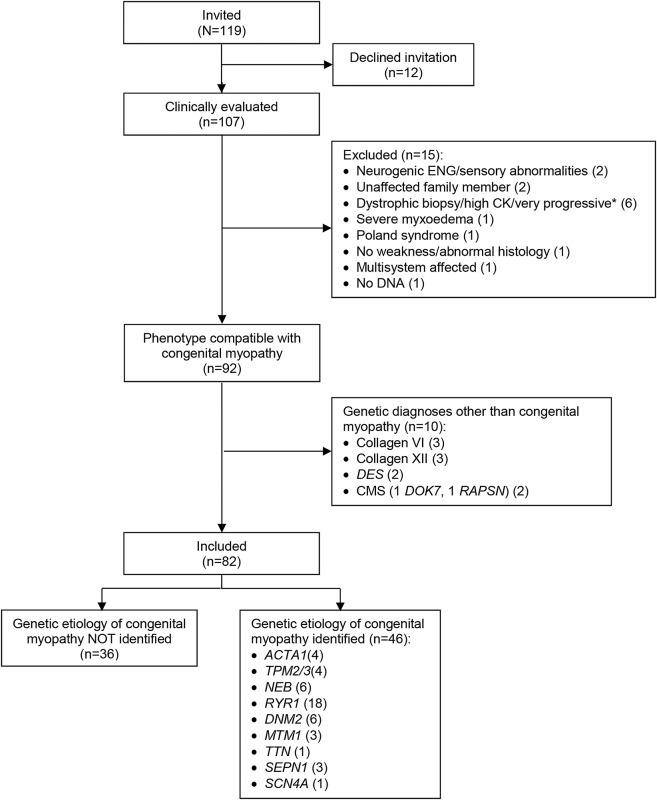
Methods: A national cohort of 107 patients ≥5 years diagnosed with congenital myopathy were prospectively assessed clinically, histologically, and genetically.
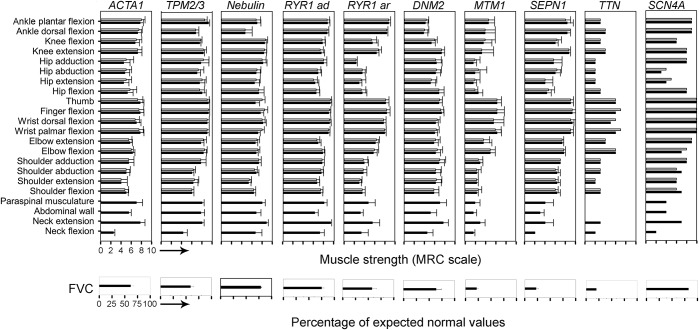
Results: Twenty-five patients were excluded because of atypical features or alternative etiologies. The remaining 82 were on average 28 years old. Histologic examination revealed 14 (17%) with core disease, 15 (18%) centronuclear myopathy, 12 (15%) nemaline rods, 27 (33%) congenital fiber-type disproportion or type I predominance, and 14 (17%) nonspecific myopathic changes. Genetic etiology was identified in 46 patients (56.1%); 22.0% were heterozygous or compound heterozygous for mutations in RYR1, 7.3% had DNM2 mutations, and 7.3% NEB mutations. Less than 5% had mutations in ACTA1, TPM2/3, MTM1, TTN, SEPN1, or SC4NA. A genetic cause was established in 83% with specific histology (cores/rods/centronuclear myopathy) vs 29% with unspecific histology. The detailed clinical examination found gene-dependent discrepancies in the pattern of muscle affection and walking ability. Although walking ability was delayed in patients with ACTA1, TPM2/3, and RYR1 mutations, it was within normal limits in patients with NEB and DNM2 mutations.
Conclusions: We found that overall, genetic and histologic prevalence of congenital myopathy in Denmark differs from previous retrospective reports. Less RYR1 and more DNM2 and NEB mutations and less core histology were present in our cohort. These differences may be explained by our prospective design, the older cohort of patients, and by differences in genetic background.
Figures
References
-
- Maggi L, Scoto M, Cirak S, et al. Congenital myopathies—clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul Disord 2013;23:195–205. - PubMed
-
- Brandt A, Schleithoff L, Jurkat-Rott K, Klingler W, Baur C, Lehmann-Horn F. Screening of the ryanodine receptor gene in 105 malignant hyperthermia families: novel mutations and concordance with the in vitro contracture test. Hum Mol Genet 1999;8:2055–2062. - PubMed
-
- Carmignac V, Salih MA, Quijano-Roy S, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 2007;61:340–351. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous