

Review
doi: 10.1038/nature21701.
Exploiting non-covalent π interactions for catalyst design
Affiliations
- PMID: 28358089
- PMCID: PMC5907483
- DOI: 10.1038/nature21701
Item in Clipboard
Review
Exploiting non-covalent π interactions for catalyst design
Nature.
.
Abstract
Molecular recognition, binding and catalysis are often mediated by non-covalent interactions involving aromatic functional groups. Although the relative complexity of these so-called π interactions has made them challenging to study, theory and modelling have now reached the stage at which we can explain their physical origins and obtain reliable insight into their effects on molecular binding and chemical transformations. This offers opportunities for the rational manipulation of these complex non-covalent interactions and their direct incorporation into the design of small-molecule catalysts and enzymes.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
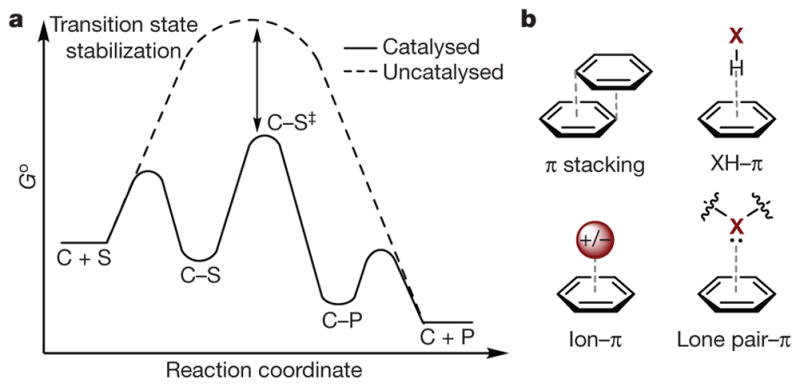
a, Qualitative depiction of catalysis via transition-state (TS) stabilization, where C is catalyst, S is substrate, and P is product. Go, Gibbs free energy in standard state; reaction coordinate indicates the progression of the reaction; C–S‡, activated complex. b, Featured interactions (grey dashed lines) of π systems in this Review. X = B, C, N, O.
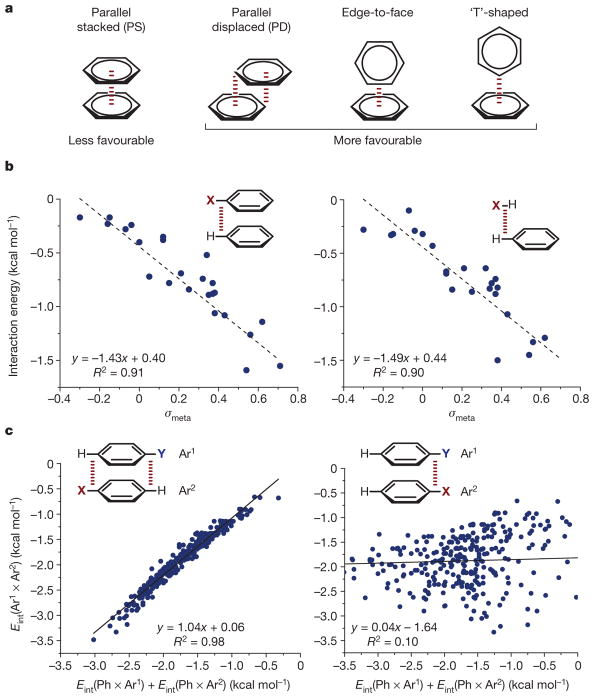
a, π–π interaction geometries (see text for details). b, Hammett correlations (interaction energy versus σmeta, the meta-Hammett substituent parameter) supporting the direct interaction model. Broken red line in insets indicates interaction. Adapted from ref. , American Chemical Society. c, Correlations demonstrating geometric consequences of the direct interaction model. Insets show Ar1 (top) and Ar2 (bottom); broken red lines in insets indicate interactions. On axes, Eint(Ar1 × Ar2) denotes interaction energy between Ar1 and Ar2, and so on. Adapted from ref. , American Chemical Society. In b and c, regression lines and their equations are shown: R, correlation coefficient.
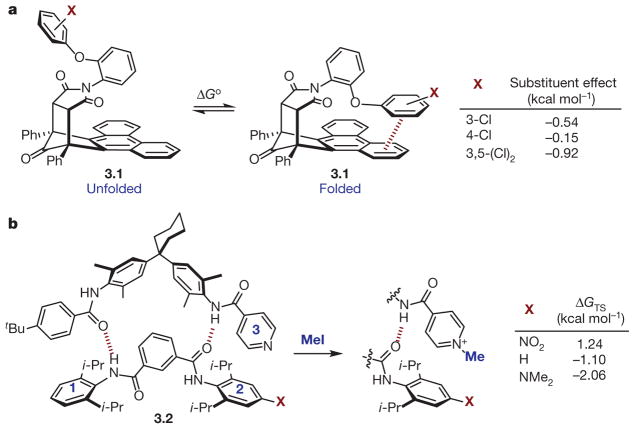
a, Substituent effects on π-stacking interactions detected using molecular torsion balance 3.1 (shown unfolded and folded); Δ Go = –RTlnKeq. b, Transition-state interaction energies (Δ GTS) of rings 2 and 3 during conversion of pyridine of 3.2 (left) to methyl pyridinium (right) using methyl iodide (MeI).
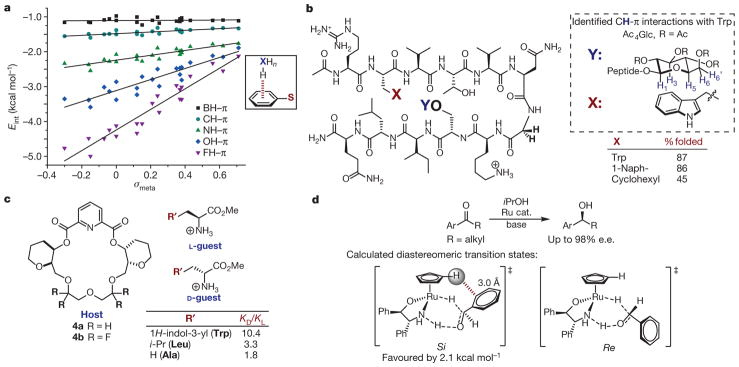
a, Effect of X and arene identity on XH–π interactions. The structure that was computed to determine Eint in each case is shown boxed at right; S is a variable substituent. Adapted from ref. , American Chemical Society. b, Double mutant cycles in β-hairpin oligopeptide to quantify CH–π interactions. Left, the oligopeptide under evaluation, where X and Y are variable substituents. Previous studies support the CH–π interactions as shown in the box on the right. c, Chiral host (left) binds amino acid guests (right) selectively through a CH–π interaction, as measured by KD/KL. d, Rationalization of enantioselectivity in asymmetric transfer hydrogenation of aryl ketones. Top, reaction studied; bottom, calculated diastereomeric transition states (Si and Re isomers shown left and right, respectively). e.e., enantiomeric excess.
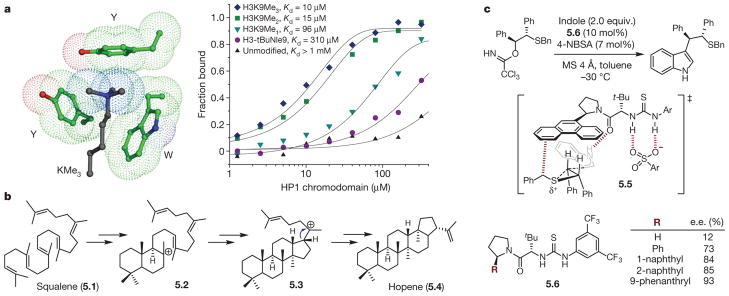
a, Left, depiction of the aromatic box in the HP1 chromodomain binding trimethyllysine (KMe3) of histone 3 (H3: Y, tyrosine; W, tryptophan). Green, HP1 chromodomain carbon; blue, nitrogen; red, oxygen; black, H3 carbon. Right, binding of HP1 chromodomain to wild-type H3 (unmodified) and a series of mutants (tBuNle9, tert-butyl norleucine; K9Me1, methyllysine; K9Me2, dimethyllysine; K9Me3 trimethyllysine). Reprinted from ref. , National Academy of Sciences, USA. b, Proposed mechanism for hopene synthesis from squalene. c, Nucleophilic ring opening of episulfonium ions is stabilized by a cation–π interaction: top, reaction; middle, transition state; bottom, effect of catalyst substituents on product enantiomeric excess.
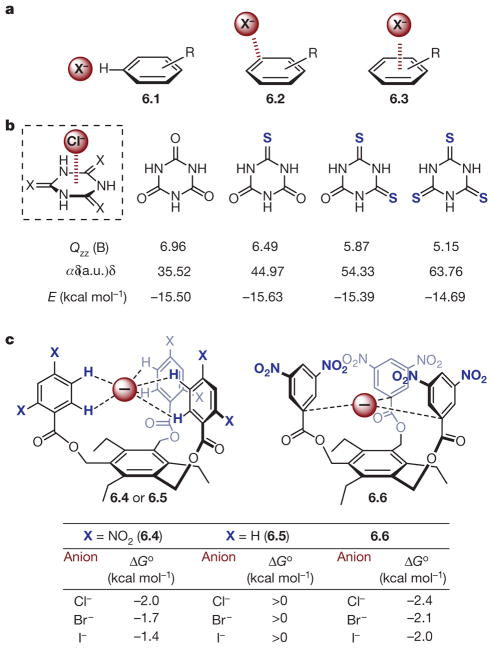
a, Anion–π geometries exemplified in 6.1, 6.2 and 6.3. b, Effect of polarizability on anion–π interaction. Boxed structure, interaction geometry. Values of parameters are shown under the relevant structures: Qzz, z2 component of the quadrupole moment tensor; α, molecular polarizability; E, energy. c, Neutral receptors for anion binding. Top, depiction of bimolecular association between neutral receptors 6.4–6.6 and halide anions Cl−, Br− and I−; bottom, the free energies of association (Δ Go).
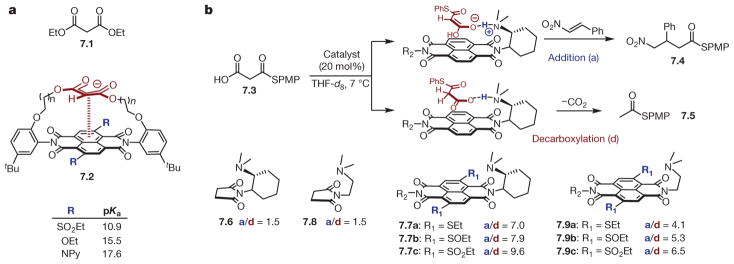
a, Modulation of pKa values of malonate moiety embedded within naphthalenediimide (NDI) scaffold (7.2, bottom) through anion–π interactions compared with diethylmalonate as a control (7.1, top). b, Top, depiction of differential stabilization of planar (leading to addition product 7.4) and non-planar (leading to decarboxylation product 7.5) tautomers of malonic acid half thioester (7.3) by NDI scaffold. Bottom, effect of modulation of NDI (7.7 and 7.9) electronic properties on product distribution (addition versus decarboxylation, a/d) and comparison with controls (7.6 and 7.8) incapable participating in anion–π interactions. Differential transition-state stabilization is tuned over a range of 1.3 kcal mol−1.
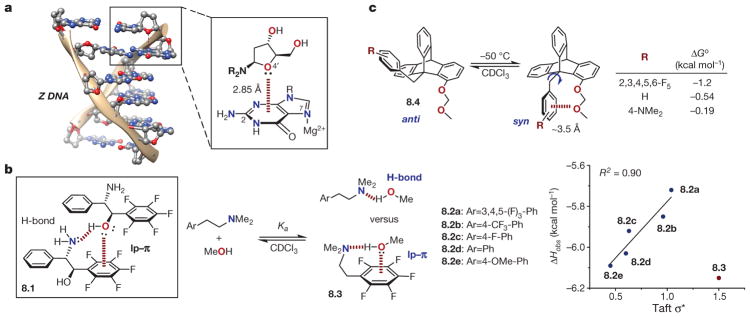
a, Example of lone pair(lp)–π interaction in Z-DNA (broken red line in magnified inset at right). Adapted from ref. , PCCP Owner Societies, and refs and , American Chemical Society. b, Quantification of lp–π interaction in solution. Box at left, depiction of the dimeric solid-state structure of amino alcohol 8.1 implicating lp–π interactions as important structural elements. Middle, association between electronically diverse dimethylamino arylethylamines (8.2 and 8.3) and methanol in solution, and (right) correlation between enthalpy of association (Δ Hobs) and the Taft parameter (σ* ). c, Quantification of lp–π interactions using molecular torsion balances (anti and syn isomers of 8.4). Δ Go = –RTlnKeq, where Keq = [syn]/[anti].
References
-
- Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34:939–945. - PubMed
-
- Kirby AJ. Enzyme mechanisms, models, and mimics. Angew Chem Int Edn Engl. 1996;35:706–724.
-
- Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. - PubMed
-
- Biedermann F, Schneider HJ. Experimental binding energies in supramolecular complexes. Chem Rev. 2016;116:5216–5300. - PubMed
-
- Schneider HJ. Binding mechanisms in supramolecular complexes. Angew Chem Int Ed. 2009;48:3924–3977. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
