

Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells
- PMID: 28358377
- PMCID: PMC5386536
- DOI: 10.1038/cddis.2017.133
Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells
Abstract
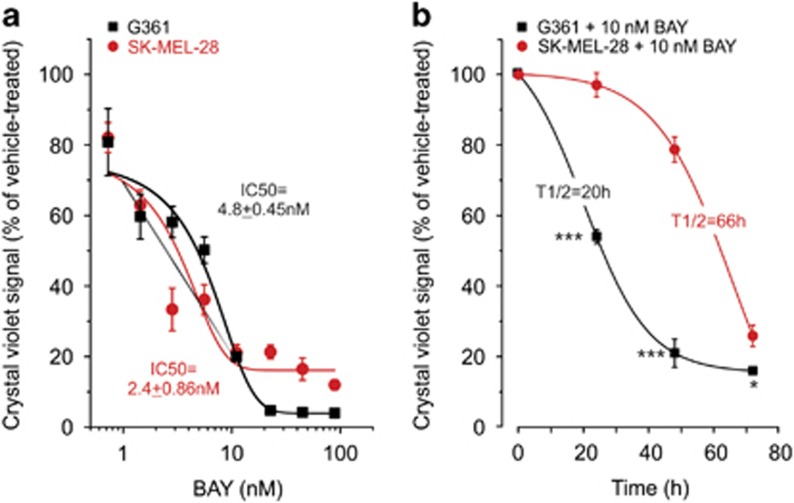
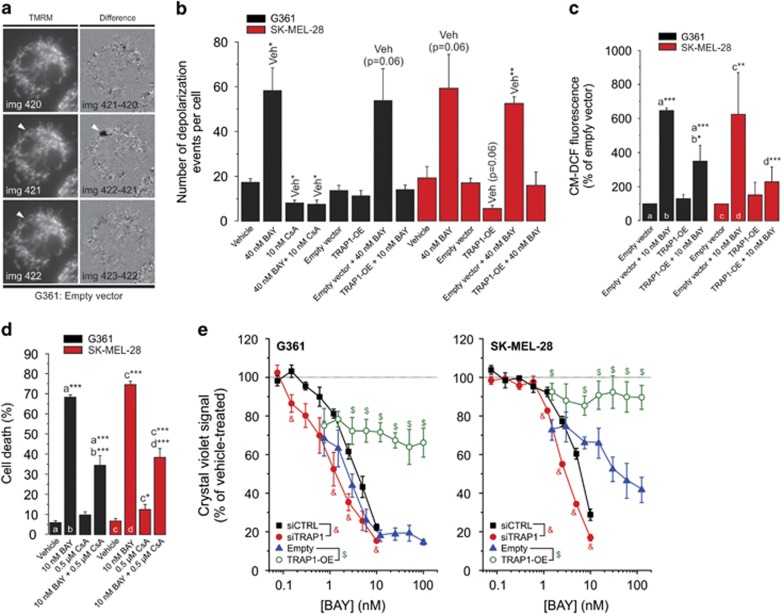
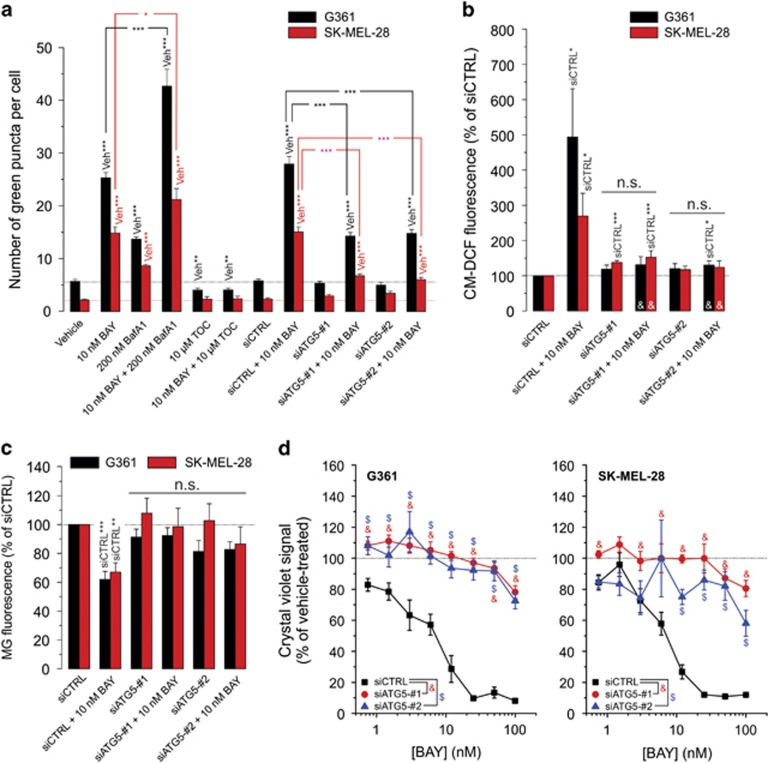
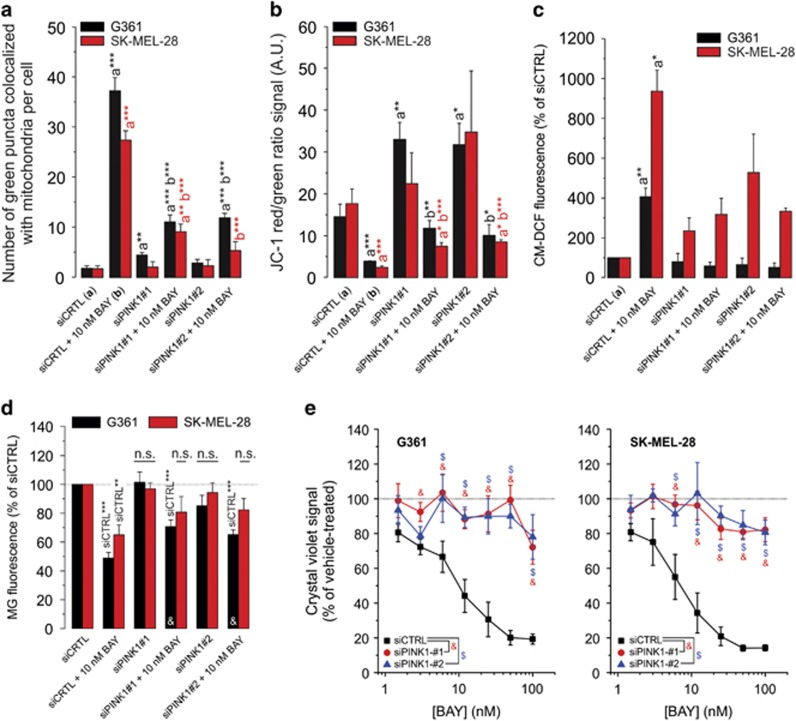
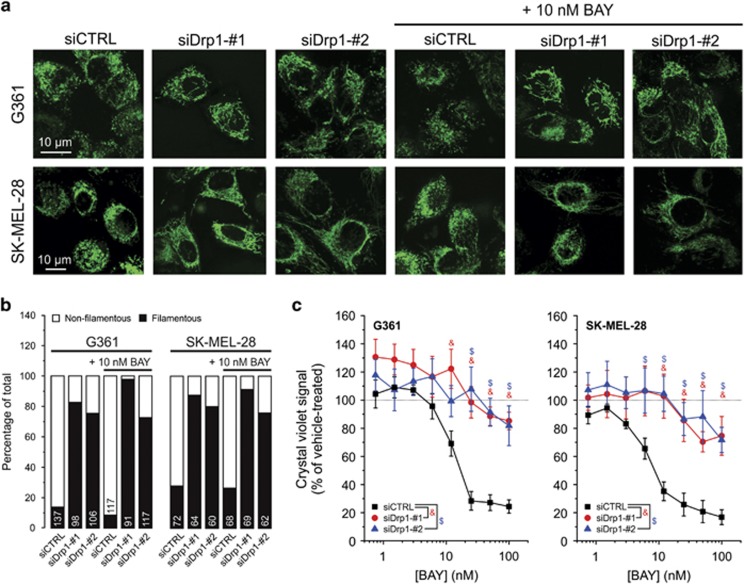
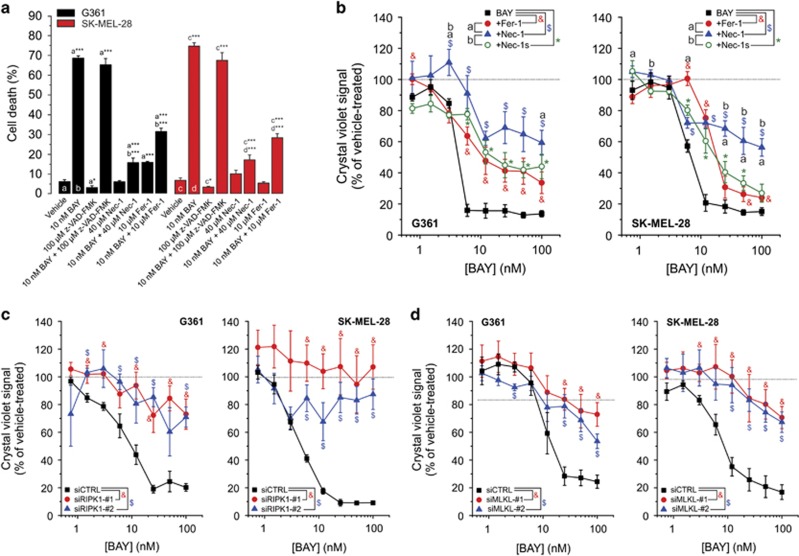
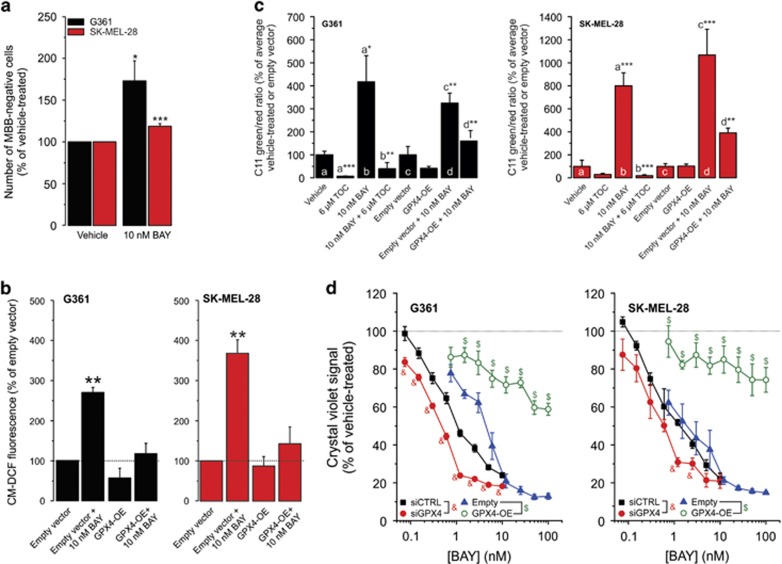
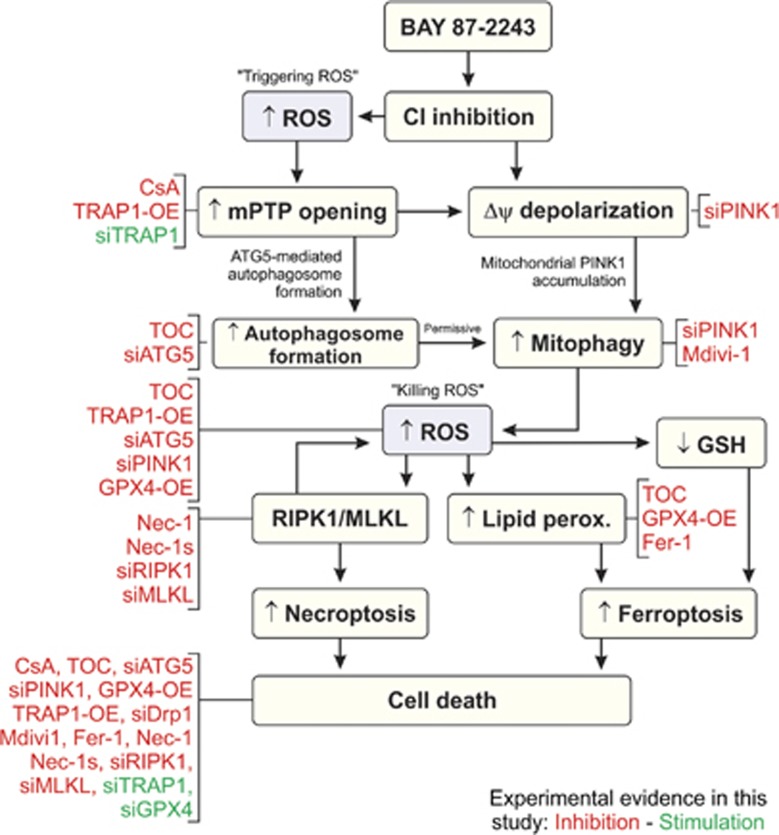
Inhibition of complex I (CI) of the mitochondrial respiratory chain by BAY 87-2243 ('BAY') triggers death of BRAFV600E melanoma cell lines and inhibits in vivo tumor growth. Here we studied the mechanism by which this inhibition induces melanoma cell death. BAY treatment depolarized the mitochondrial membrane potential (Δψ), increased cellular ROS levels, stimulated lipid peroxidation and reduced glutathione levels. These effects were paralleled by increased opening of the mitochondrial permeability transition pore (mPTP) and stimulation of autophagosome formation and mitophagy. BAY-induced cell death was not due to glucose shortage and inhibited by the antioxidant α-tocopherol and the mPTP inhibitor cyclosporin A. Tumor necrosis factor receptor-associated protein 1 (TRAP1) overexpression in BAY-treated cells lowered ROS levels and inhibited mPTP opening and cell death, whereas the latter was potentiated by TRAP1 knockdown. Knockdown of autophagy-related 5 (ATG5) inhibited the BAY-stimulated autophagosome formation, cellular ROS increase and cell death. Knockdown of phosphatase and tensin homolog-induced putative kinase 1 (PINK1) inhibited the BAY-induced Δψ depolarization, mitophagy stimulation, ROS increase and cell death. Dynamin-related protein 1 (Drp1) knockdown induced mitochondrial filamentation and inhibited BAY-induced cell death. The latter was insensitive to the pancaspase inhibitor z-VAD-FMK, but reduced by necroptosis inhibitors (necrostatin-1, necrostatin-1s)) and knockdown of key necroptosis proteins (receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and mixed lineage kinase domain-like (MLKL)). BAY-induced cell death was also reduced by the ferroptosis inhibitor ferrostatin-1 and overexpression of the ferroptosis-inhibiting protein glutathione peroxidase 4 (GPX4). This overexpression also inhibited the BAY-induced ROS increase and lipid peroxidation. Conversely, GPX4 knockdown potentiated BAY-induced cell death. We propose a chain of events in which: (i) CI inhibition induces mPTP opening and Δψ depolarization, that (ii) stimulate autophagosome formation, mitophagy and an associated ROS increase, leading to (iii) activation of combined necroptotic/ferroptotic cell death.
Conflict of interest statement
FB, LMPEvO, HMB, SEvEdV, JCWH, SG, PHGMW and WJHK declare no competing interests. LS, CK and MH are full-time employees of Bayer Pharma AG, which developed BAY 87-2243.
Figures
References
-
- Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. TR Mol Med 2015; 21: 164–171. - PubMed
-
- Shakya A, Cooksey R, Cox JE, Wang V, McClain DA, Tantin D. Oct1 loss of function induces a coordinate metabolic shift that opposes tumorigenicity. Nat Cell Biol 2009; 11: 320–327. - PubMed
-
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 2004; 64: 985–993. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
