

Glucocorticoids Induce Bone and Muscle Atrophy by Tissue-Specific Mechanisms Upstream of E3 Ubiquitin Ligases
- PMID: 28359087
- PMCID: PMC5460781
- DOI: 10.1210/en.2016-1779
Glucocorticoids Induce Bone and Muscle Atrophy by Tissue-Specific Mechanisms Upstream of E3 Ubiquitin Ligases
Abstract
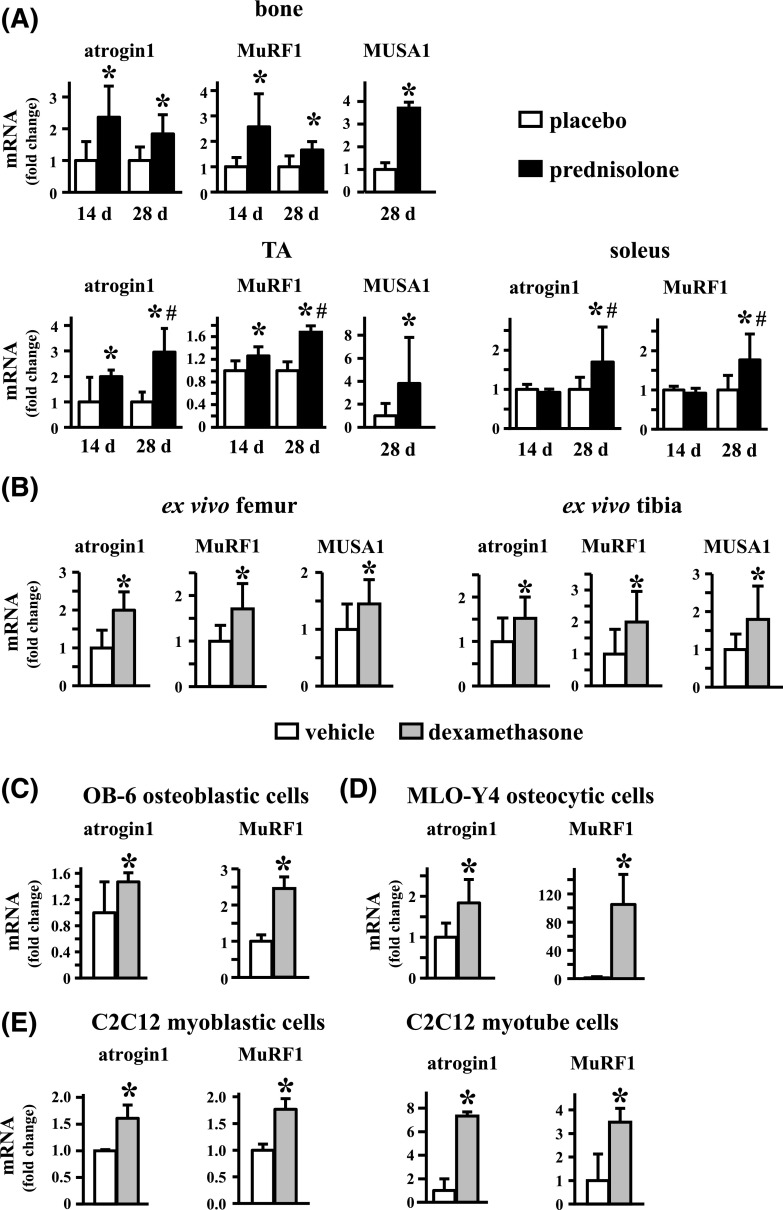
Glucocorticoid excess, either endogenous with diseases of the adrenal gland, stress, or aging or when administered for immunosuppression, induces bone and muscle loss, leading to osteopenia and sarcopenia. Muscle weakness increases the propensity for falling, which, combined with the lower bone mass, increases the fracture risk. The mechanisms underlying glucocorticoid-induced bone and muscle atrophy are not completely understood. We have demonstrated that the loss of bone and muscle mass, decreased bone formation, and reduced muscle strength, hallmarks of glucocorticoid excess, are accompanied by upregulation in both tissues in vivo of the atrophy-related genes atrogin1, MuRF1, and MUSA1. These are E3 ubiquitin ligases traditionally considered muscle-specific. Glucocorticoids also upregulated atrophy genes in cultured osteoblastic/osteocytic cells, in ex vivo bone organ cultures, and in muscle organ cultures and C2C12 myoblasts/myotubes. Furthermore, glucocorticoids markedly increased the expression of components of the Notch signaling pathway in muscle in vivo, ex vivo, and in vitro. In contrast, glucocorticoids did not increase Notch signaling in bone or bone cells. Moreover, the increased expression of atrophy-related genes in muscle, but not in bone, and the decreased myotube diameter induced by glucocorticoids were prevented by inhibiting Notch signaling. Thus, glucocorticoids activate different mechanisms in bone and muscle that upregulate atrophy-related genes. However, the role of these genes in the effects of glucocorticoids in bone is unknown. Nevertheless, these findings advance our knowledge of the mechanism of action of glucocorticoids in the musculoskeletal system and provide the basis for novel therapies to prevent glucocorticoid-induced atrophy of bone and muscle.
Copyright © 2017 by the Endocrine Society.
Figures






References
-
- Weinstein RS. Clinical practice: glucocorticoid-induced bone disease. N Engl J Med. 2011;365(1):62–70. - PubMed
-
- Rizzoli R, Adachi JD, Cooper C, Dere W, Devogelaer JP, Diez-Perez A, Kanis JA, Laslop A, Mitlak B, Papapoulos S, Ralston S, Reiter S, Werhya G, Reginster JY. Management of glucocorticoid-induced osteoporosis. Calcif Tissue Int. 2012;91(4):225–243. - PubMed
-
- Reid IR. Glucocorticoid osteoporosis—mechanisms and management. Eur J Endocrinol. 1997;137(3):209–217. - PubMed
-
- Suzuki Y, Nawata H, Soen S, Fujiwara S, Nakayama H, Tanaka I, Ozono K, Sagawa A, Takayanagi R, Tanaka H, Miki T, Masunari N, Tanaka Y. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research: 2014 update. J Bone Miner Metab. 2014;32(4):337–350. - PubMed
-
- van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int. 2002;13(10):777–787. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
