

Tropomyosin 1: Multiple roles in the developing heart and in the formation of congenital heart defects
- PMID: 28359939
- PMCID: PMC5441184
- DOI: 10.1016/j.yjmcc.2017.03.006
Tropomyosin 1: Multiple roles in the developing heart and in the formation of congenital heart defects
Abstract
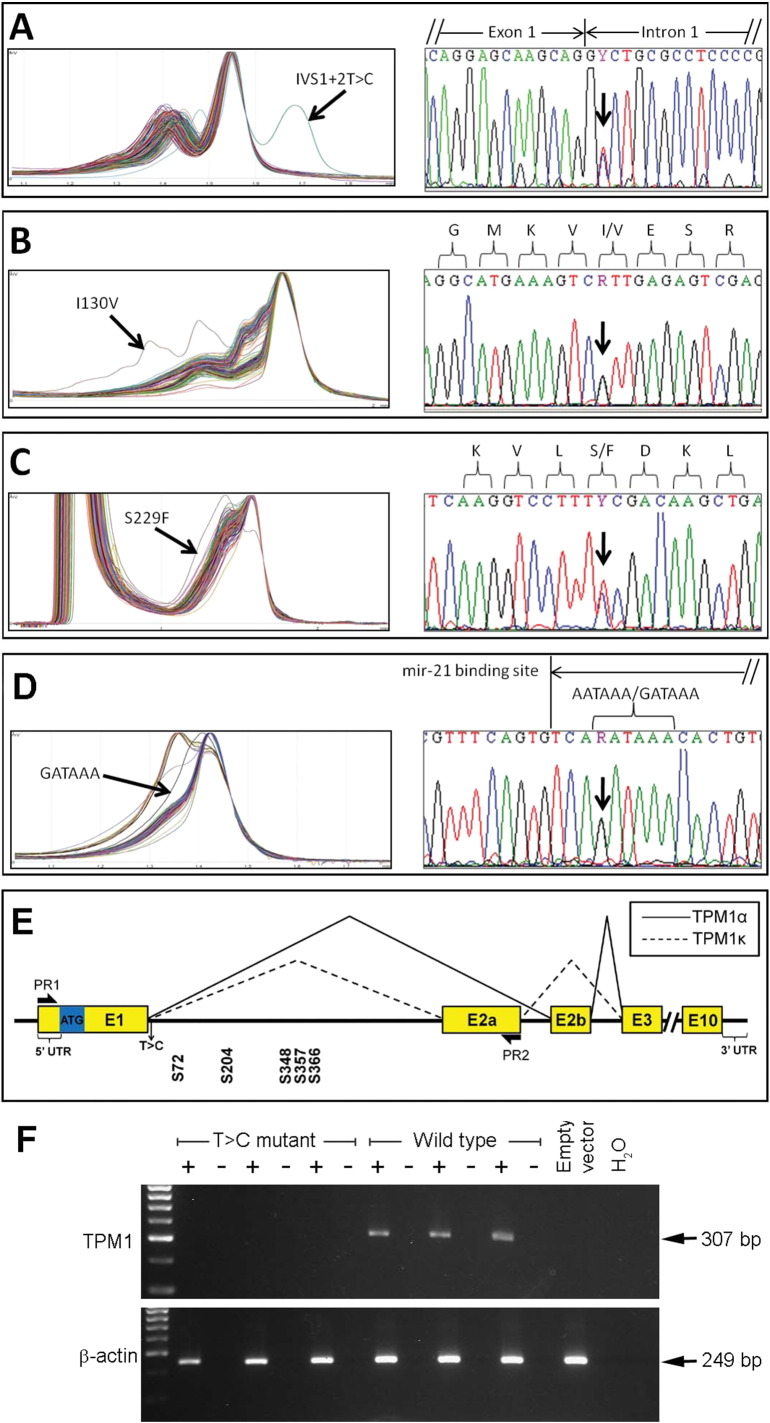
Tropomyosin 1 (TPM1) is an essential sarcomeric component, stabilising the thin filament and facilitating actin's interaction with myosin. A number of sarcomeric proteins, such as alpha myosin heavy chain, play crucial roles in cardiac development. Mutations in these genes have been linked to congenital heart defects (CHDs), occurring in approximately 1 in 145 live births. To date, TPM1 has not been associated with isolated CHDs. Analysis of 380 CHD cases revealed three novel mutations in the TPM1 gene; IVS1+2T>C, I130V, S229F and a polyadenylation signal site variant GATAAA/AATAAA. Analysis of IVS1+2T>C revealed aberrant pre-mRNA splicing. In addition, abnormal structural properties were found in hearts transfected with TPM1 carrying I130V and S229F mutations. Phenotypic analysis of TPM1 morpholino-treated embryos revealed roles for TPM1 in cardiac looping, atrial septation and ventricular trabeculae formation and increased apoptosis was seen within the heart. In addition, sarcomere assembly was affected and altered action potentials were exhibited. This study demonstrated that sarcomeric TPM1 plays vital roles in cardiogenesis and is a suitable candidate gene for screening individuals with isolated CHDs.
Keywords: Cardiac development; Congenital heart defects; Structural protein; Tropomyosin 1.
Copyright © 2017 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures







Similar articles
-
Cardiac alpha-myosin (MYH6) is the predominant sarcomeric disease gene for familial atrial septal defects.PLoS One. 2011;6(12):e28872. doi: 10.1371/journal.pone.0028872. Epub 2011 Dec 14. PLoS One. 2011. PMID: 22194935 Free PMC article.
-
Sarcomere mutations in cardiogenesis and ventricular noncompaction.Trends Cardiovasc Med. 2009 Jan;19(1):17-21. doi: 10.1016/j.tcm.2009.03.003. Trends Cardiovasc Med. 2009. PMID: 19467449 Review.
-
Rescue of high expression beta-tropomyosin transgenic mice by 5-propyl-2-thiouracil. Regulating the alpha-myosin heavy chain promoter.J Biol Chem. 1999 Oct 8;274(41):29558-63. doi: 10.1074/jbc.274.41.29558. J Biol Chem. 1999. PMID: 10506222
-
[Mutations in genes for sarcomeric proteins].Nihon Rinsho. 2000 Jan;58(1):117-22. Nihon Rinsho. 2000. PMID: 10885298 Review. Japanese.
-
Coexistence of Digenic Mutations in Both Thin (TPM1) and Thick (MYH7) Filaments of Sarcomeric Genes Leads to Severe Hypertrophic Cardiomyopathy in a South Indian FHCM.DNA Cell Biol. 2015 May;34(5):350-9. doi: 10.1089/dna.2014.2650. Epub 2015 Jan 21. DNA Cell Biol. 2015. PMID: 25607779
Cited by
-
RBFOX2 is critical for maintaining alternative polyadenylation patterns and mitochondrial health in rat myoblasts.Cell Rep. 2021 Nov 2;37(5):109910. doi: 10.1016/j.celrep.2021.109910. Cell Rep. 2021. PMID: 34731606 Free PMC article.
-
Comparison of Human Tissue Microarray to Human Pericyte Transcriptome Yields Novel Perivascular Cell Markers.Stem Cells Dev. 2019 Sep 15;28(18):1214-1223. doi: 10.1089/scd.2019.0106. Epub 2019 Aug 1. Stem Cells Dev. 2019. PMID: 31264500 Free PMC article.
-
Mutations in RNA Methyltransferase Gene NSUN5 Confer High Risk of Outflow Tract Malformation.Front Cell Dev Biol. 2021 Apr 21;9:623394. doi: 10.3389/fcell.2021.623394. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33968922 Free PMC article.
-
Molecular Characterization of Tropomyosin and Its Potential Involvement in Muscle Contraction in Pacific Abalone.Genes (Basel). 2022 Dec 20;14(1):2. doi: 10.3390/genes14010002. Genes (Basel). 2022. PMID: 36672743 Free PMC article.
-
Genetic predisposition study of heart failure and its association with cardiomyopathy.Egypt Heart J. 2022 Jan 21;74(1):5. doi: 10.1186/s43044-022-00240-6. Egypt Heart J. 2022. PMID: 35061126 Free PMC article. Review.
References
-
- Hamburger V., Hamilton H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951;88:49–92. - PubMed
-
- Sissman N.J. Developmental landmarks in cardiac morphogenesis: comparative chronology. Am. J. Cardiol. 1970;25:141–148. - PubMed
-
- Morse D.E. Scanning electron microscopy of the developing septa in the chick heart. Birth Defects Orig. Artic. Ser. 1978;14:91–107. - PubMed
-
- Bentham J., Bhattacharya S. Genetic mechanisms controlling cardiovascular development. Ann. N. Y. Acad. Sci. 2008;1123:10–19. - PubMed
-
- Ching Y.H., Ghosh T.K., Cross S.J., Packham E.A., Honeyman L., Loughna S. Mutation in myosin heavy chain 6 causes atrial septal defect. Nat. Genet. 2005;37:423–428. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
