

Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications
- PMID: 28360231
- PMCID: PMC5411765
- DOI: 10.1101/gr.215095.116
Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications
Abstract
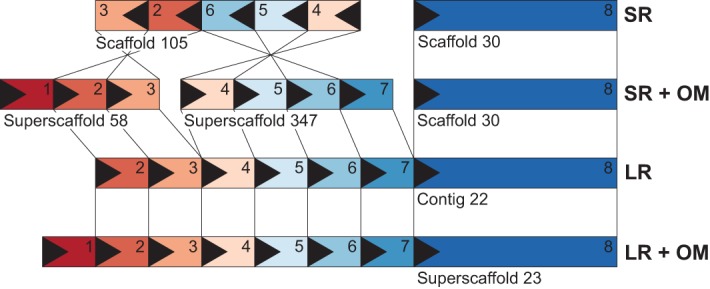
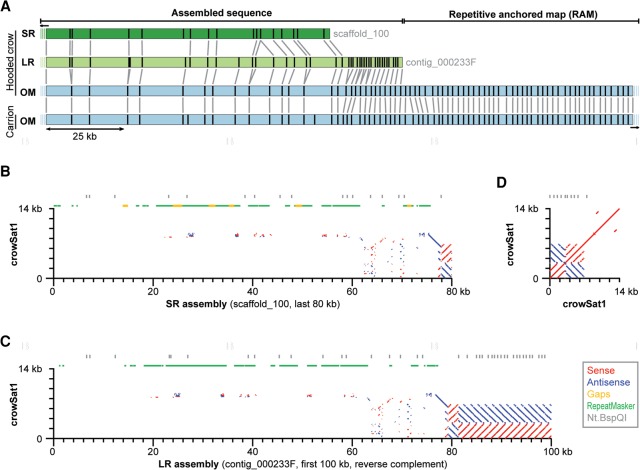
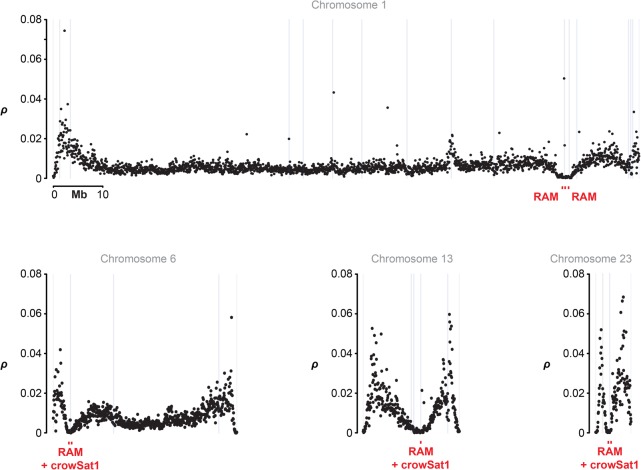
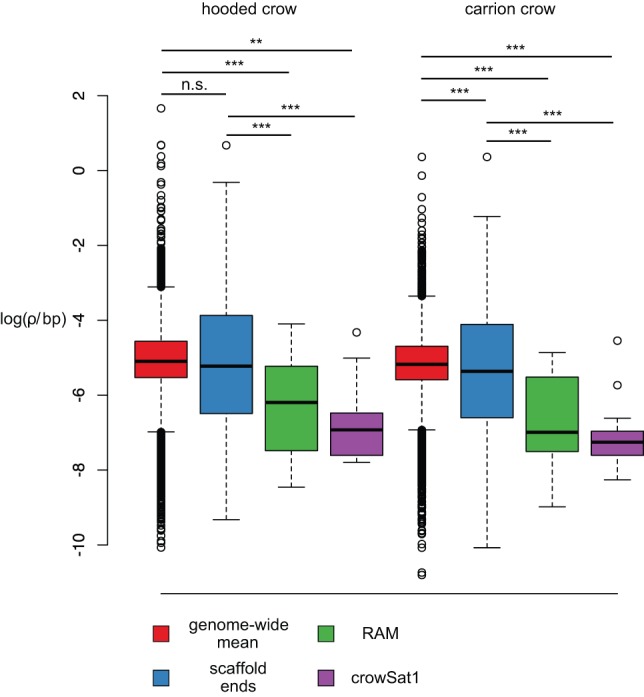
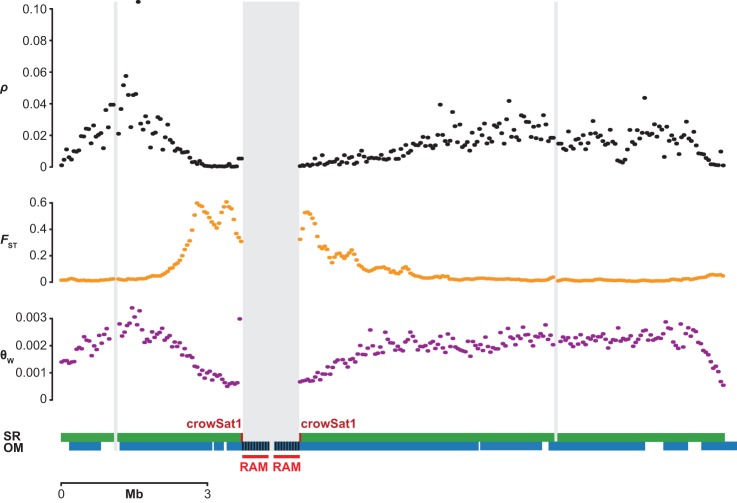
Accurate and contiguous genome assembly is key to a comprehensive understanding of the processes shaping genomic diversity and evolution. Yet, it is frequently constrained by constitutive heterochromatin, usually characterized by highly repetitive DNA. As a key feature of genome architecture associated with centromeric and subtelomeric regions, it locally influences meiotic recombination. In this study, we assess the impact of large tandem repeat arrays on the recombination rate landscape in an avian speciation model, the Eurasian crow. We assembled two high-quality genome references using single-molecule real-time sequencing (long-read assembly [LR]) and single-molecule optical maps (optical map assembly [OM]). A three-way comparison including the published short-read assembly (SR) constructed for the same individual allowed assessing assembly properties and pinpointing misassemblies. By combining information from all three assemblies, we characterized 36 previously unidentified large repetitive regions in the proximity of sequence assembly breakpoints, the majority of which contained complex arrays of a 14-kb satellite repeat or its 1.2-kb subunit. Using whole-genome population resequencing data, we estimated the population-scaled recombination rate (ρ) and found it to be significantly reduced in these regions. These findings are consistent with an effect of low recombination in regions adjacent to centromeric or subtelomeric heterochromatin and add to our understanding of the processes generating widespread heterogeneity in genetic diversity and differentiation along the genome. By combining three different technologies, our results highlight the importance of adding a layer of information on genome structure that is inaccessible to each approach independently.
© 2017 Weissensteiner et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. - PubMed
-
- Anantharaman T, Mishra B. 2001. False positives in genomic map assembly and sequence validation. In Algorithms in bioinformatics first international workshop, WABI 2001, Århus, Denmark.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials