

A computational method for estimating the PCR duplication rate in DNA and RNA-seq experiments
- PMID: 28361665
- PMCID: PMC5374682
- DOI: 10.1186/s12859-017-1471-9
A computational method for estimating the PCR duplication rate in DNA and RNA-seq experiments
Abstract
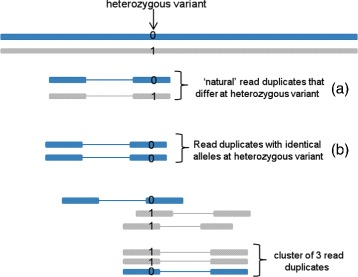
Background: PCR amplification is an important step in the preparation of DNA sequencing libraries prior to high-throughput sequencing. PCR amplification introduces redundant reads in the sequence data and estimating the PCR duplication rate is important to assess the frequency of such reads. Existing computational methods do not distinguish PCR duplicates from "natural" read duplicates that represent independent DNA fragments and therefore, over-estimate the PCR duplication rate for DNA-seq and RNA-seq experiments.
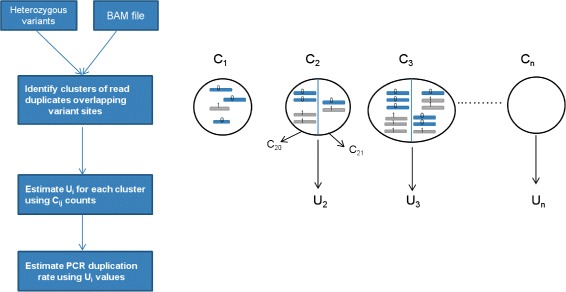
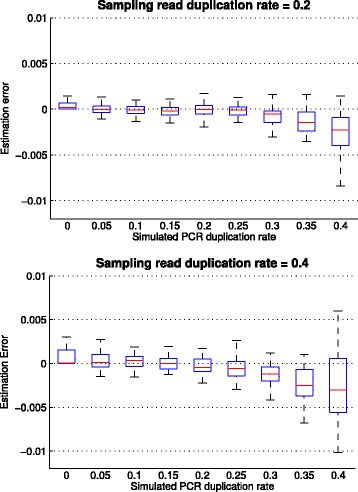
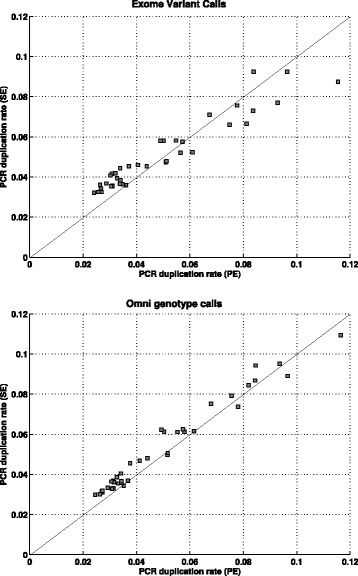
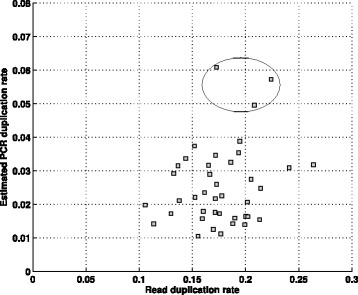
Results: In this paper, we present a computational method to estimate the average PCR duplication rate of high-throughput sequence datasets that accounts for natural read duplicates by leveraging heterozygous variants in an individual genome. Analysis of simulated data and exome sequence data from the 1000 Genomes project demonstrated that our method can accurately estimate the PCR duplication rate on paired-end as well as single-end read datasets which contain a high proportion of natural read duplicates. Further, analysis of exome datasets prepared using the Nextera library preparation method indicated that 45-50% of read duplicates correspond to natural read duplicates likely due to fragmentation bias. Finally, analysis of RNA-seq datasets from individuals in the 1000 Genomes project demonstrated that 70-95% of read duplicates observed in such datasets correspond to natural duplicates sampled from genes with high expression and identified outlier samples with a 2-fold greater PCR duplication rate than other samples.
Conclusions: The method described here is a useful tool for estimating the PCR duplication rate of high-throughput sequence datasets and for assessing the fraction of read duplicates that correspond to natural read duplicates. An implementation of the method is available at https://github.com/vibansal/PCRduplicates .
Keywords: Heterozygosity; High-throughput sequencing; Mathematical modeling; Natural duplicates; PCR duplicates; RNA-seq.
Figures
References
-
- Quail MA, Swerdlow H, Turner DJ. Improved protocols for the illumina genome analyzer sequencing system. Curr Protoc Hum Genet. 2009; Chapter 18: Unit 18.2. http://dx.doi.org/10.1002/0471142905.hg1802s62. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
