

AfterQC: automatic filtering, trimming, error removing and quality control for fastq data
- PMID: 28361673
- PMCID: PMC5374548
- DOI: 10.1186/s12859-017-1469-3
AfterQC: automatic filtering, trimming, error removing and quality control for fastq data
Abstract
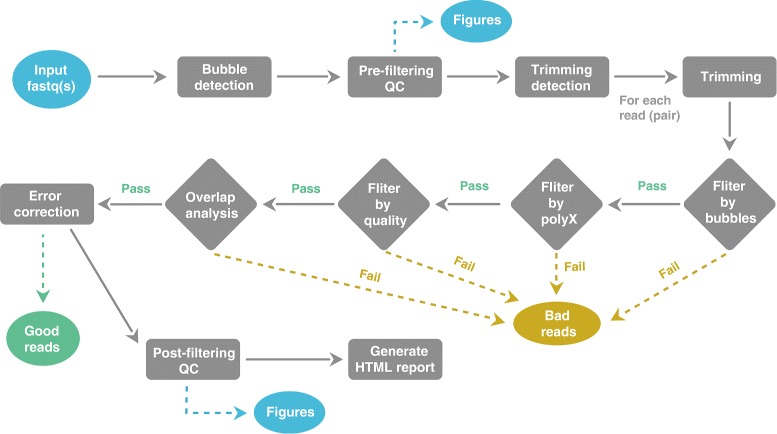
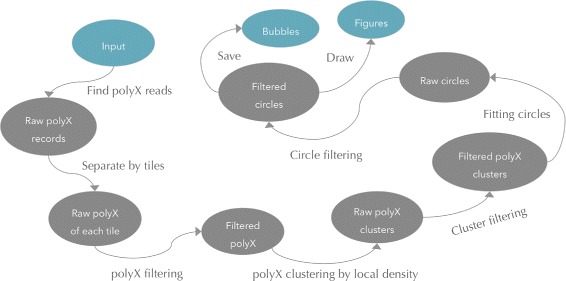
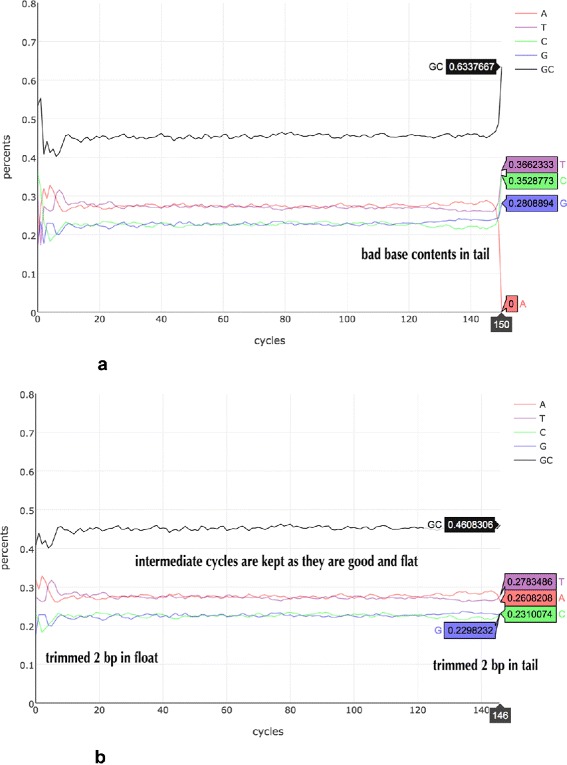
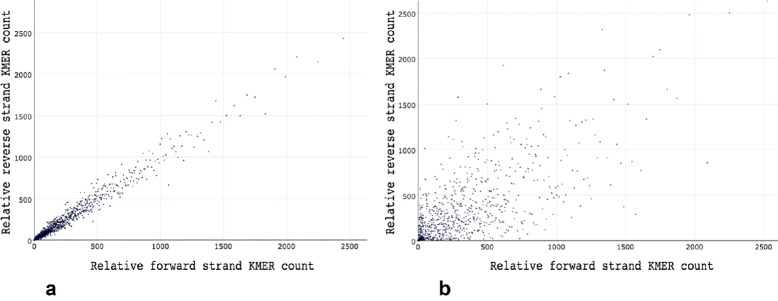
Background: Some applications, especially those clinical applications requiring high accuracy of sequencing data, usually have to face the troubles caused by unavoidable sequencing errors. Several tools have been proposed to profile the sequencing quality, but few of them can quantify or correct the sequencing errors. This unmet requirement motivated us to develop AfterQC, a tool with functions to profile sequencing errors and correct most of them, plus highly automated quality control and data filtering features. Different from most tools, AfterQC analyses the overlapping of paired sequences for pair-end sequencing data. Based on overlapping analysis, AfterQC can detect and cut adapters, and furthermore it gives a novel function to correct wrong bases in the overlapping regions. Another new feature is to detect and visualise sequencing bubbles, which can be commonly found on the flowcell lanes and may raise sequencing errors. Besides normal per cycle quality and base content plotting, AfterQC also provides features like polyX (a long sub-sequence of a same base X) filtering, automatic trimming and K-MER based strand bias profiling.
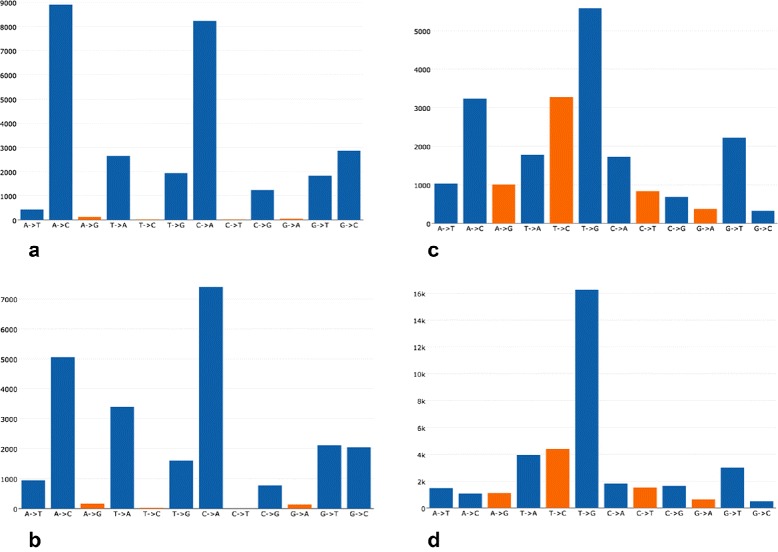
Results: For each single or pair of FastQ files, AfterQC filters out bad reads, detects and eliminates sequencer's bubble effects, trims reads at front and tail, detects the sequencing errors and corrects part of them, and finally outputs clean data and generates HTML reports with interactive figures. AfterQC can run in batch mode with multiprocess support, it can run with a single FastQ file, a single pair of FastQ files (for pair-end sequencing), or a folder for all included FastQ files to be processed automatically. Based on overlapping analysis, AfterQC can estimate the sequencing error rate and profile the error transform distribution. The results of our error profiling tests show that the error distribution is highly platform dependent.
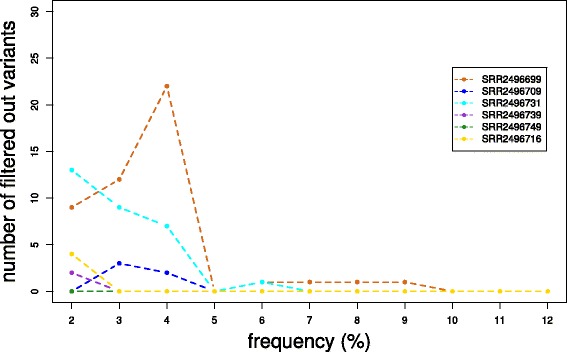
Conclusion: Much more than just another new quality control (QC) tool, AfterQC is able to perform quality control, data filtering, error profiling and base correction automatically. Experimental results show that AfterQC can help to eliminate the sequencing errors for pair-end sequencing data to provide much cleaner outputs, and consequently help to reduce the false-positive variants, especially for the low-frequency somatic mutations. While providing rich configurable options, AfterQC can detect and set all the options automatically and require no argument in most cases.
Keywords: Bubble; Data filtering; NGS; Overlap analysis; Quality control.
Figures



References
-
- Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. A tale of three next generation sequencing platforms: comparison of ion torrent, pacific biosciences and illumina miseq sequencers. BMC genomics. 2012;13(1):1. doi: 10.1186/1471-2164-13-341. - DOI - PMC - PubMed
-
- Andrews S. A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/. Accessed 7 Dec 2016.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
