

BATVI: Fast, sensitive and accurate detection of virus integrations
- PMID: 28361674
- PMCID: PMC5374687
- DOI: 10.1186/s12859-017-1470-x
BATVI: Fast, sensitive and accurate detection of virus integrations
Abstract
Background: The study of virus integrations in human genome is important since virus integrations were shown to be associated with diseases. In the literature, few methods have been proposed that predict virus integrations using next generation sequencing datasets. Although they work, they are slow and are not very sensitive.
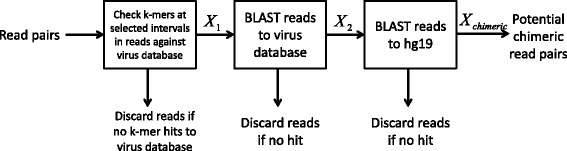
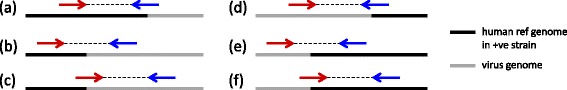
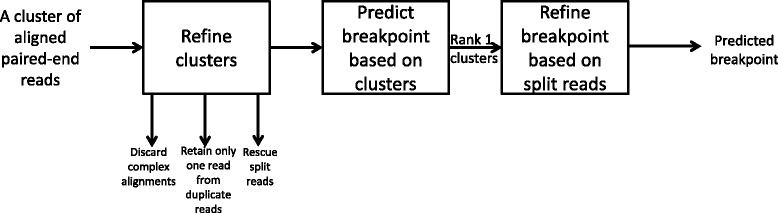
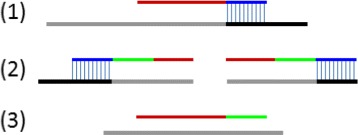
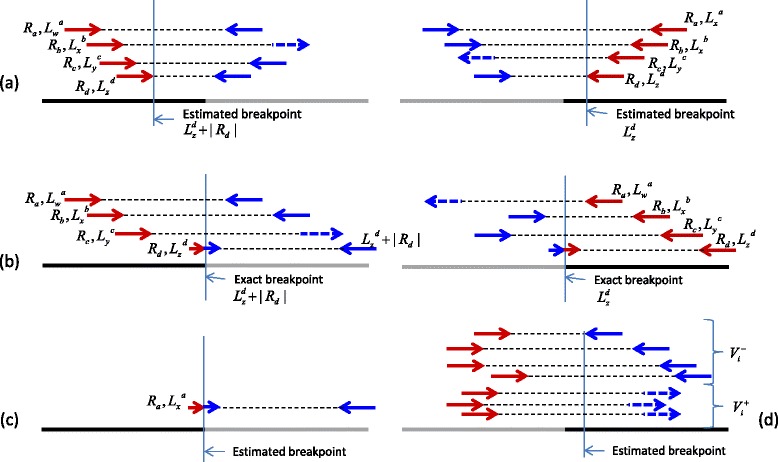
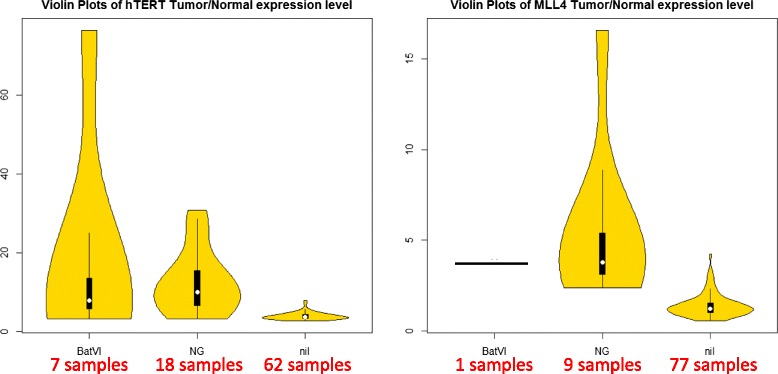
Results and discussion: This paper introduces a new method BatVI to predict viral integrations. Our method uses a fast screening method to filter out chimeric reads containing possible viral integrations. Next, sensitive alignments of these candidate chimeric reads are called by BLAST. Chimeric reads that are co-localized in the human genome are clustered. Finally, by assembling the chimeric reads in each cluster, high confident virus integration sites are extracted.
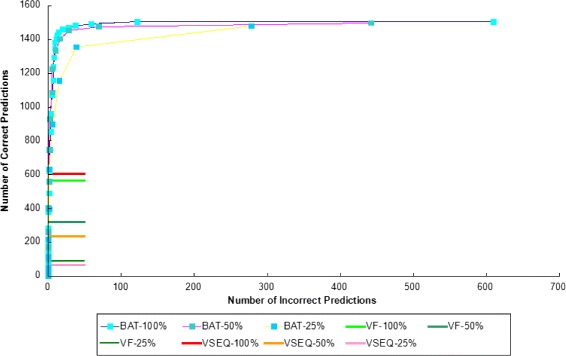
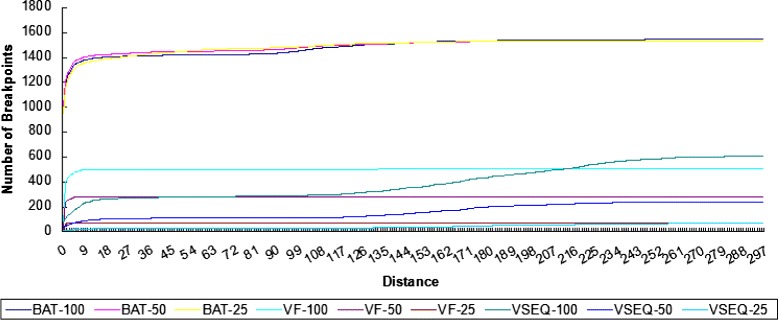
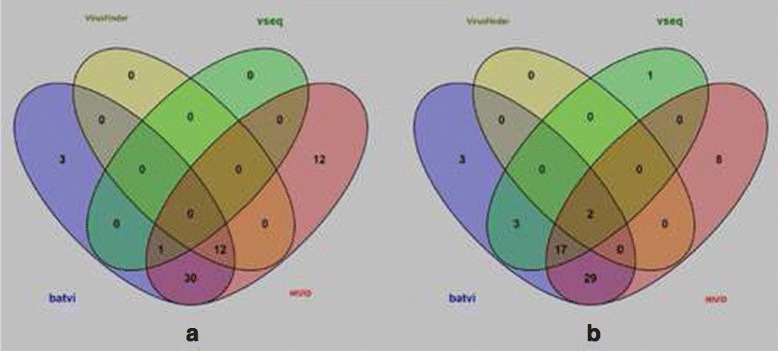
Conclusion: We compared the performance of BatVI with existing methods VirusFinder and VirusSeq using both simulated and real-life datasets of liver cancer patients. BatVI ran an order of magnitude faster and was able to predict almost twice the number of true positives compared to other methods while maintaining a false positive rate less than 1%. For the liver cancer datasets, BatVI uncovered novel integrations to two important genes TERT and MLL4, which were missed by previous studies. Through gene expression data, we verified the correctness of these additional integrations. BatVI can be downloaded from http://biogpu.ddns.comp.nus.edu.sg/~ksung/batvi/index.html .
Keywords: Alignment; NGS; Viral integration.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
