

Pysim-sv: a package for simulating structural variation data with GC-biases
- PMID: 28361688
- PMCID: PMC5374556
- DOI: 10.1186/s12859-017-1464-8
Pysim-sv: a package for simulating structural variation data with GC-biases
Abstract
Background: Structural variations (SVs) are wide-spread in human genomes and may have important implications in disease-related and evolutionary studies. High-throughput sequencing (HTS) has become a major platform for SV detection and simulation serves as a powerful and cost-effective approach for benchmarking SV detection algorithms. Accurate performance assessment by simulation requires the simulator capable of generating simulation data with all important features of real data, such GC biases in HTS data and various complexities in tumor data. However, no available package has systematically addressed all issues in data simulation for SV benchmarking.
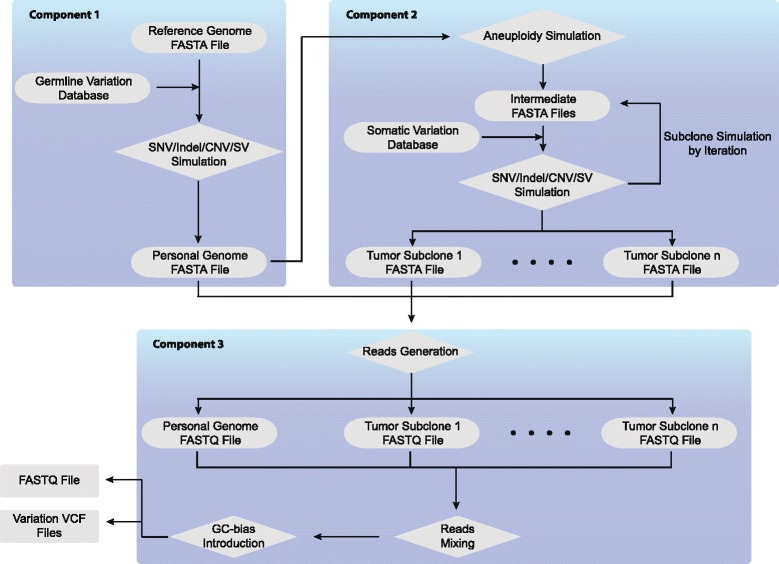
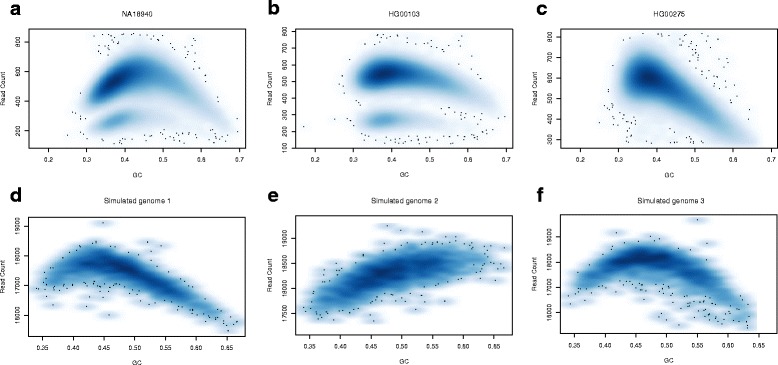
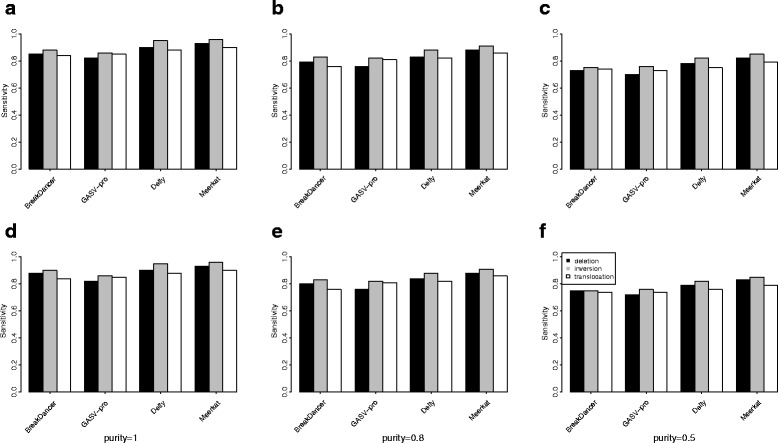
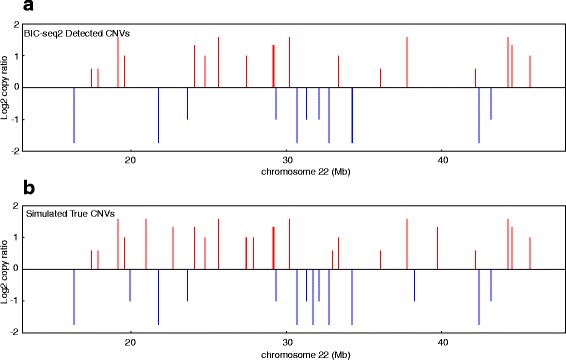
Results: Pysim-sv is a package for simulating HTS data to evaluate performance of SV detection algorithms. Pysim-sv can introduce a wide spectrum of germline and somatic genomic variations. The package contains functionalities to simulate tumor data with aneuploidy and heterogeneous subclones, which is very useful in assessing algorithm performance in tumor studies. Furthermore, Pysim-sv can introduce GC-bias, the most important and prevalent bias in HTS data, in the simulated HTS data.
Conclusions: Pysim-sv provides an unbiased toolkit for evaluating HTS-based SV detection algorithms.
Keywords: Breakpoints; Copy number variation; Next-generation sequencing; Translocation.
Figures
References
-
- Sismani C, Koufaris C, Voskarides K. Genomic Elements in Health, Disease and Evolution. New York: Springer; 2015. Copy number variation in human health, disease and evolution.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
