

Myocardial stress and autophagy: mechanisms and potential therapies
- PMID: 28361977
- PMCID: PMC6245608
- DOI: 10.1038/nrcardio.2017.35
Myocardial stress and autophagy: mechanisms and potential therapies
Abstract
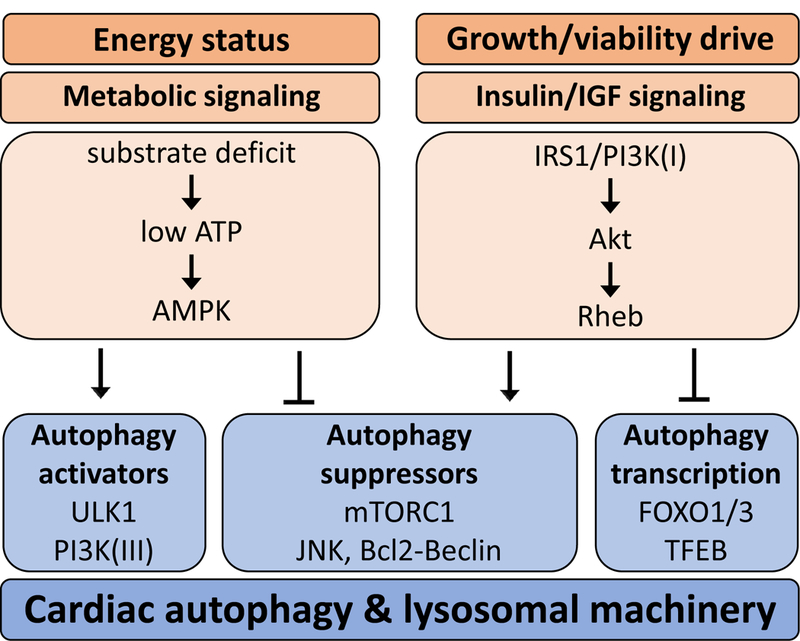
Autophagy is a ubiquitous cellular catabolic process responsive to energy stress. Research over the past decade has revealed that cardiomyocyte autophagy is a prominent homeostatic pathway, important in adaptation to altered myocardial metabolic demand. The cellular machinery of autophagy involves targeted direction of macromolecules and organelles for lysosomal degradation. Activation of autophagy has been identified as cardioprotective in some settings (that is, ischaemia and ischaemic preconditioning). In other situations, sustained autophagy has been linked with cardiopathology (for example, sustained pressure overload and heart failure). Perturbation of autophagy in diabetic cardiomyopathy has also been observed and is associated with both adaptive and maladaptive responses to stress. Emerging research findings indicate that various forms of selective autophagy operate in parallel to manage various types of catabolic cellular cargo including mitochondria, large proteins, glycogen, and stored lipids. In this Review, induction of autophagy associated with cardiac benefit or detriment is considered. The various static and dynamic approaches used to measure autophagy are critiqued, and current inconsistencies in the understanding of autophagy regulation in the heart are highlighted. The prospects for pharmacological intervention to achieve therapeutic manipulation of autophagic processes are also discussed.
Figures



References
-
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N and Otsu K The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13, 619–624 (2007). - PubMed
-
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T and Mizushima N The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036 (2004). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
