

Non-coding cancer driver candidates identified with a sample- and position-specific model of the somatic mutation rate
- PMID: 28362259
- PMCID: PMC5440169
- DOI: 10.7554/eLife.21778
Non-coding cancer driver candidates identified with a sample- and position-specific model of the somatic mutation rate
Abstract
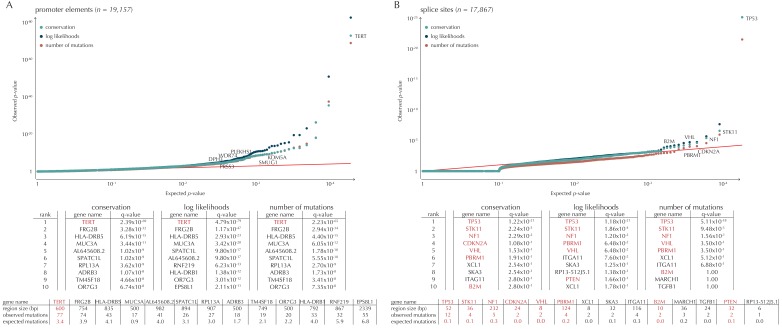
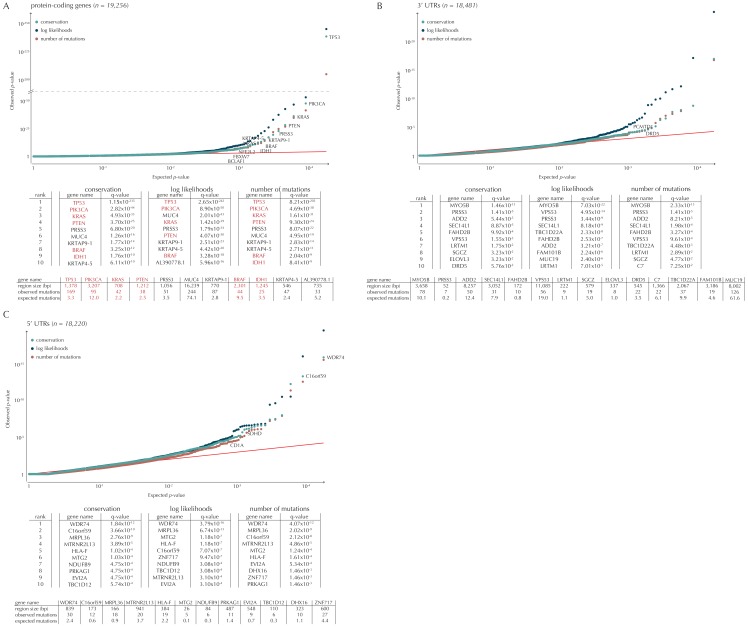
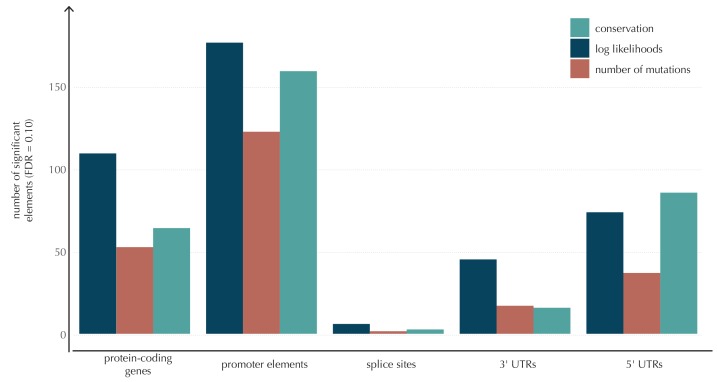
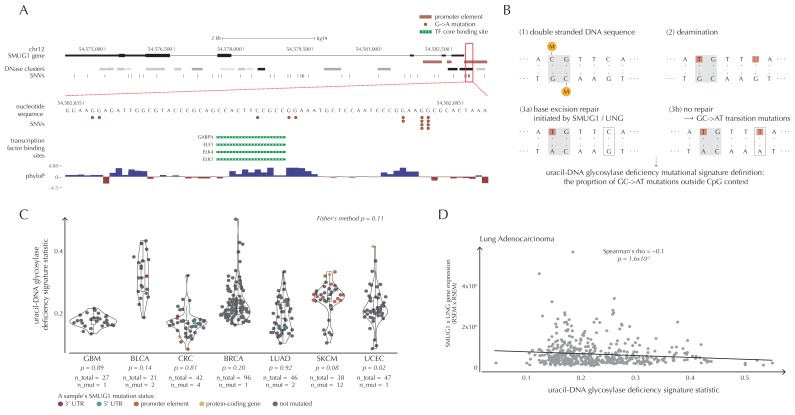
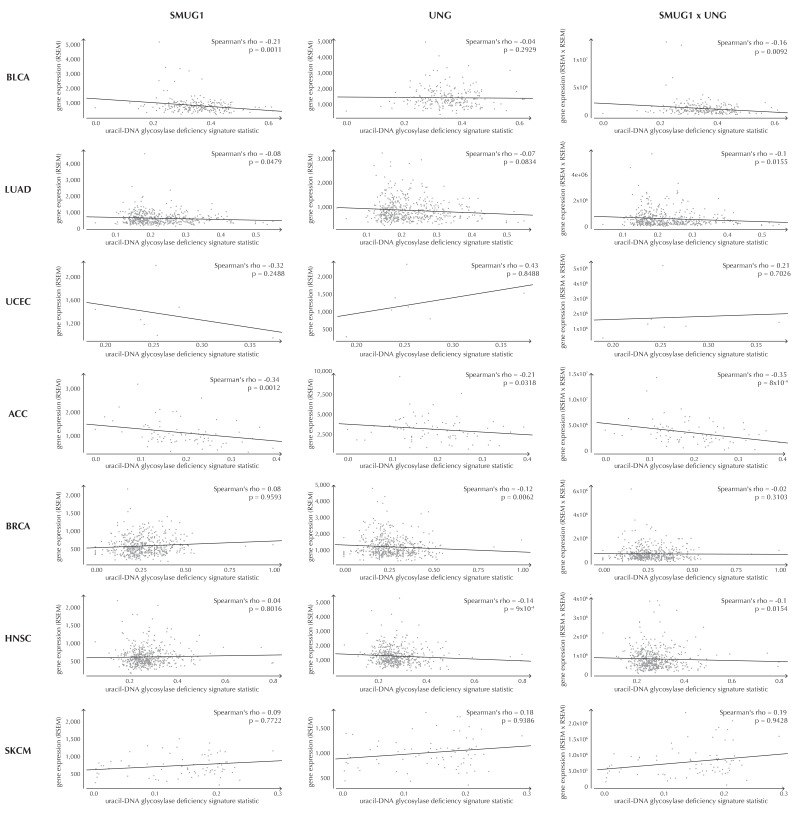
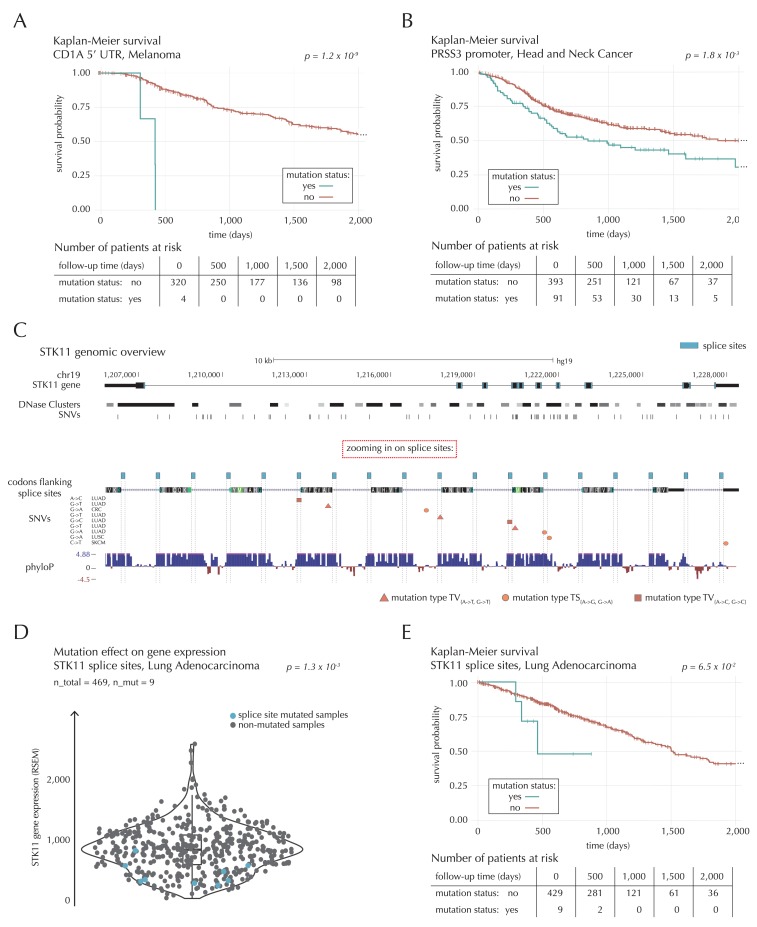
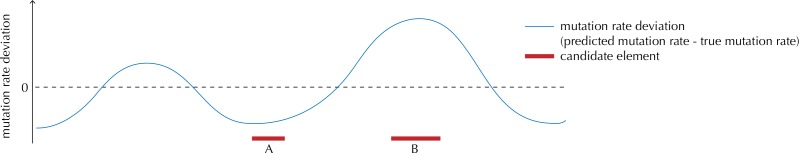
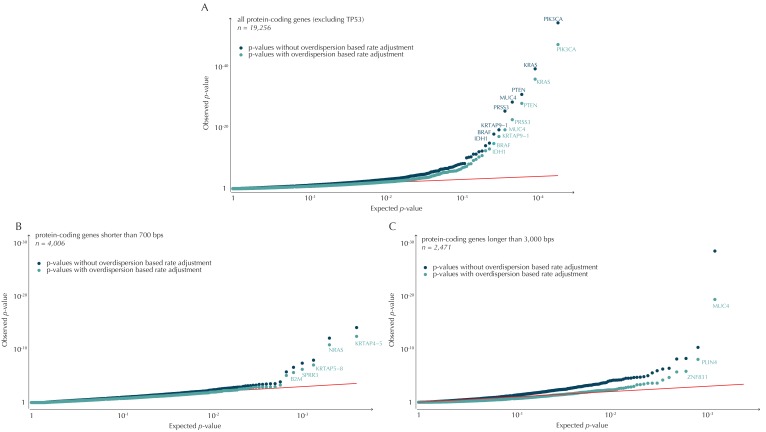
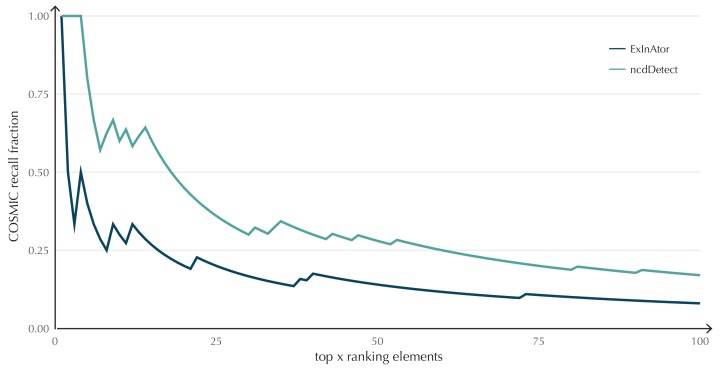
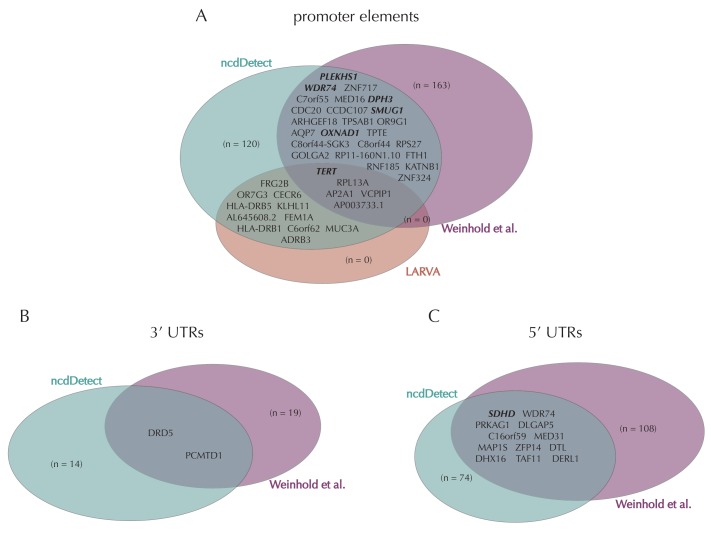
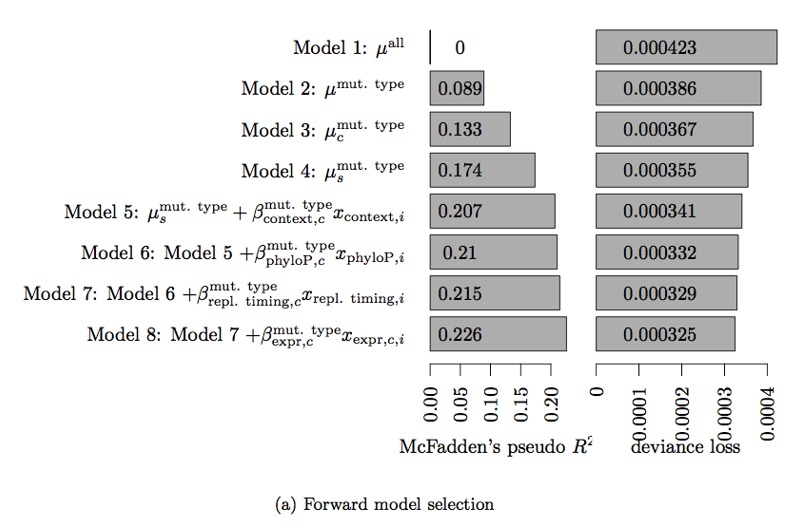
Non-coding mutations may drive cancer development. Statistical detection of non-coding driver regions is challenged by a varying mutation rate and uncertainty of functional impact. Here, we develop a statistically founded non-coding driver-detection method, ncdDetect, which includes sample-specific mutational signatures, long-range mutation rate variation, and position-specific impact measures. Using ncdDetect, we screened non-coding regulatory regions of protein-coding genes across a pan-cancer set of whole-genomes (n = 505), which top-ranked known drivers and identified new candidates. For individual candidates, presence of non-coding mutations associates with altered expression or decreased patient survival across an independent pan-cancer sample set (n = 5454). This includes an antigen-presenting gene (CD1A), where 5'UTR mutations correlate significantly with decreased survival in melanoma. Additionally, mutations in a base-excision-repair gene (SMUG1) correlate with a C-to-T mutational-signature. Overall, we find that a rich model of mutational heterogeneity facilitates non-coding driver identification and integrative analysis points to candidates of potential clinical relevance.
Keywords: cancer; cancer biology; computational biology; driver detection; human; mutational processes; non-coding mutations; systems biology.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

Similar articles
-
Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival.NPJ Genom Med. 2018 Jan 11;3:1. doi: 10.1038/s41525-017-0040-5. eCollection 2018. NPJ Genom Med. 2018. PMID: 29354286 Free PMC article.
-
Identification of coding and non-coding mutational hotspots in cancer genomes.BMC Genomics. 2017 Jan 5;18(1):17. doi: 10.1186/s12864-016-3420-9. BMC Genomics. 2017. PMID: 28056774 Free PMC article.
-
Integration of somatic mutation, expression and functional data reveals potential driver genes predictive of breast cancer survival.Bioinformatics. 2015 Aug 15;31(16):2607-13. doi: 10.1093/bioinformatics/btv164. Epub 2015 Mar 24. Bioinformatics. 2015. PMID: 25810432
-
Mining the coding and non-coding genome for cancer drivers.Cancer Lett. 2015 Dec 28;369(2):307-15. doi: 10.1016/j.canlet.2015.09.015. Epub 2015 Oct 1. Cancer Lett. 2015. PMID: 26433158 Review.
-
Finding cancer driver mutations in the era of big data research.Biophys Rev. 2019 Feb;11(1):21-29. doi: 10.1007/s12551-018-0415-6. Epub 2018 Apr 2. Biophys Rev. 2019. PMID: 29611034 Free PMC article. Review.
Cited by
-
Analyses of non-coding somatic drivers in 2,658 cancer whole genomes.Nature. 2020 Feb;578(7793):102-111. doi: 10.1038/s41586-020-1965-x. Epub 2020 Feb 5. Nature. 2020. PMID: 32025015 Free PMC article.
-
A pan-cancer atlas of cancer hallmark-associated candidate driver lncRNAs.Mol Oncol. 2018 Nov;12(11):1980-2005. doi: 10.1002/1878-0261.12381. Epub 2018 Oct 2. Mol Oncol. 2018. PMID: 30216655 Free PMC article.
-
ncdDetect2: improved models of the site-specific mutation rate in cancer and driver detection with robust significance evaluation.Bioinformatics. 2019 Jan 15;35(2):189-199. doi: 10.1093/bioinformatics/bty511. Bioinformatics. 2019. PMID: 29945188 Free PMC article.
-
Identifying somatic driver mutations in cancer with a language model of the human genome.Comput Struct Biotechnol J. 2025 Jan 17;27:531-540. doi: 10.1016/j.csbj.2025.01.011. eCollection 2025. Comput Struct Biotechnol J. 2025. PMID: 39968174 Free PMC article.
-
Identification of Cancer Drivers at CTCF Insulators in 1,962 Whole Genomes.Cell Syst. 2019 May 22;8(5):446-455.e8. doi: 10.1016/j.cels.2019.04.001. Epub 2019 May 8. Cell Syst. 2019. PMID: 31078526 Free PMC article.
References
-
- Agresti A. Categorical Data Analysis. John Wiley & Sons; 2013.
-
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Imielinsk M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Serena N-Z, Samuel AJ, Sam B, Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human Cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. - DOI - PMC - PubMed
-
- Bates D, Maechler M. MatrixModels: modelling with sparse and dense matrices. 2015 http://CRAN.R-project.org/package=MatrixModels
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
