

Facilitated dissociation of transcription factors from single DNA binding sites
- PMID: 28364020
- PMCID: PMC5402408
- DOI: 10.1073/pnas.1701884114
Facilitated dissociation of transcription factors from single DNA binding sites
Abstract
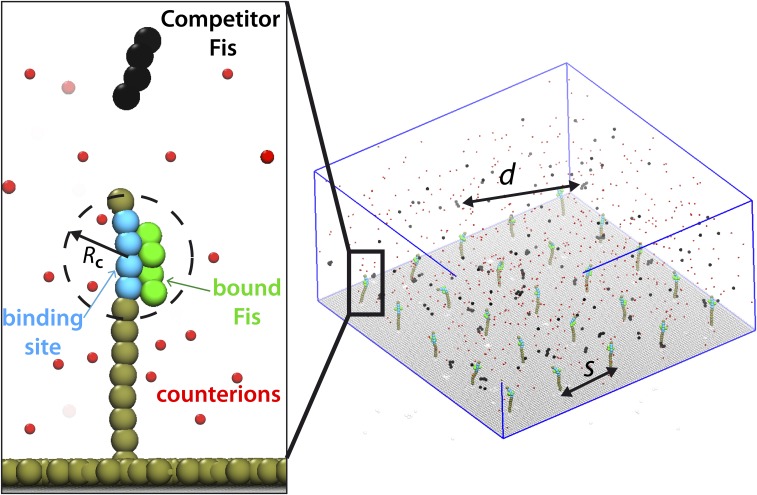
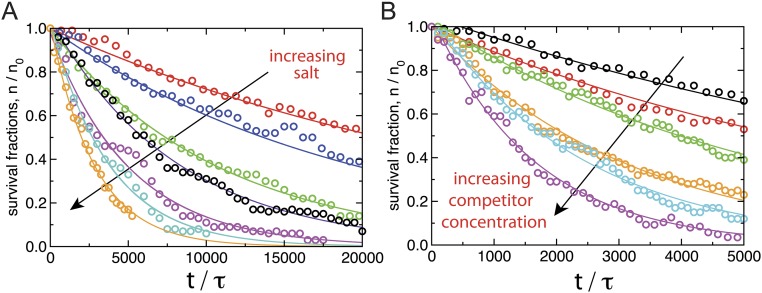
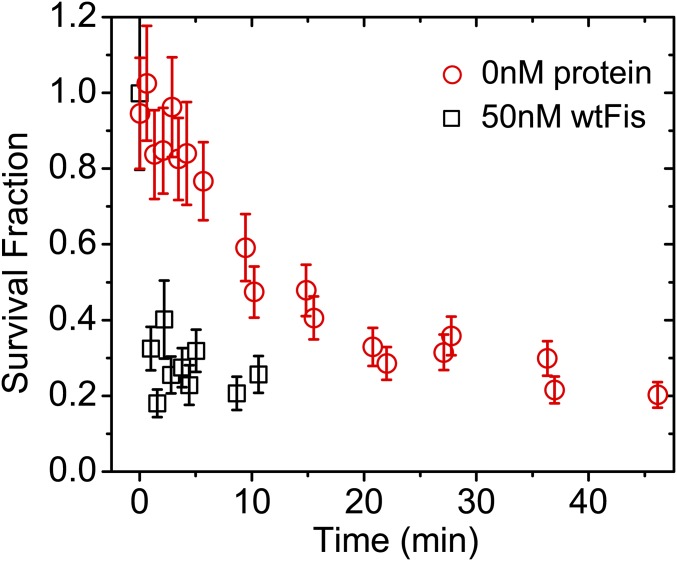
The binding of transcription factors (TFs) to DNA controls most aspects of cellular function, making the understanding of their binding kinetics imperative. The standard description of bimolecular interactions posits that TF off rates are independent of TF concentration in solution. However, recent observations have revealed that proteins in solution can accelerate the dissociation of DNA-bound proteins. To study the molecular basis of facilitated dissociation (FD), we have used single-molecule imaging to measure dissociation kinetics of Fis, a key Escherichia coli TF and major bacterial nucleoid protein, from single dsDNA binding sites. We observe a strong FD effect characterized by an exchange rate [Formula: see text], establishing that FD of Fis occurs at the single-binding site level, and we find that the off rate saturates at large Fis concentrations in solution. Although spontaneous (i.e., competitor-free) dissociation shows a strong salt dependence, we find that FD depends only weakly on salt. These results are quantitatively explained by a model in which partially dissociated bound proteins are susceptible to invasion by competitor proteins in solution. We also report FD of NHP6A, a yeast TF with structure that differs significantly from Fis. We further perform molecular dynamics simulations, which indicate that FD can occur for molecules that interact far more weakly than those that we have studied. Taken together, our results indicate that FD is a general mechanism assisting in the local removal of TFs from their binding sites and does not necessarily require cooperativity, clustering, or binding site overlap.
Keywords: DNA–protein interactions; biomolecule binding; chemical kinetics; facilitated dissociation; transcription factor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
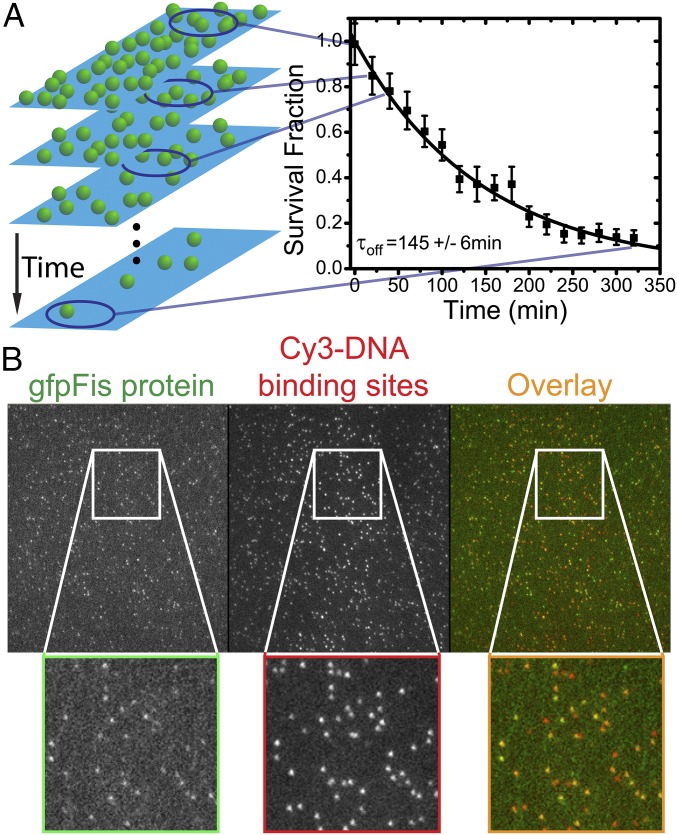
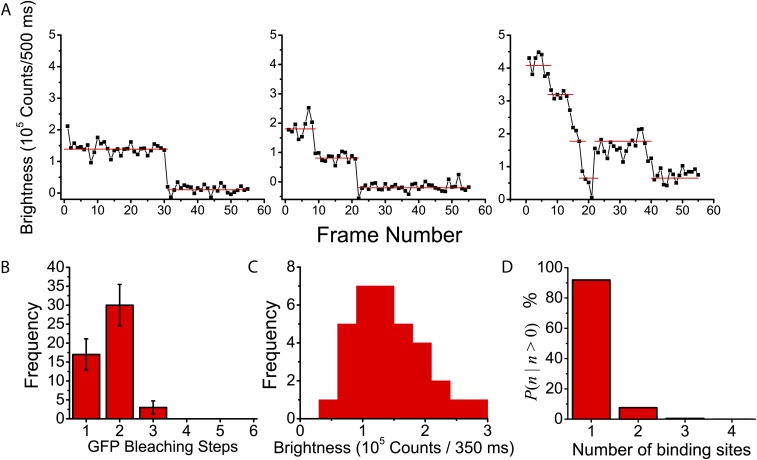
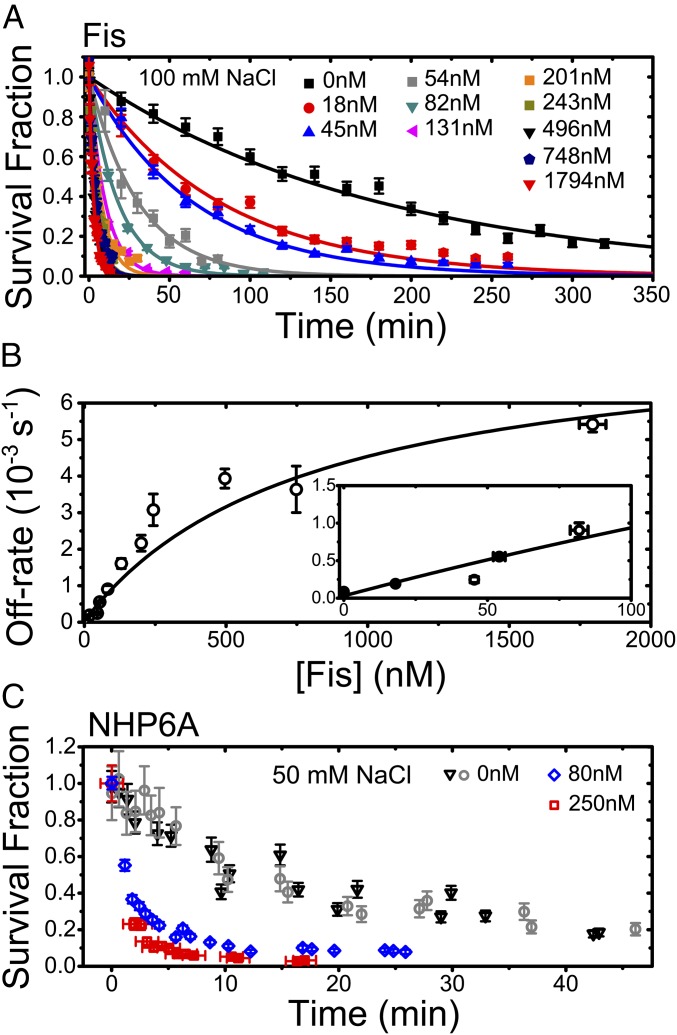
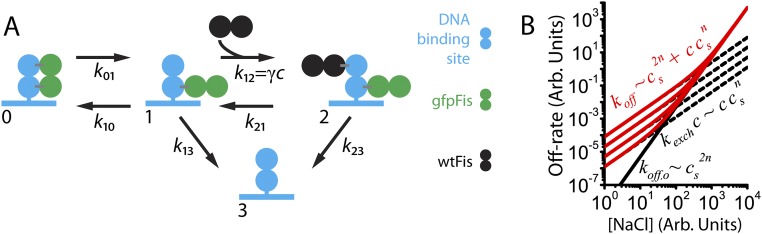
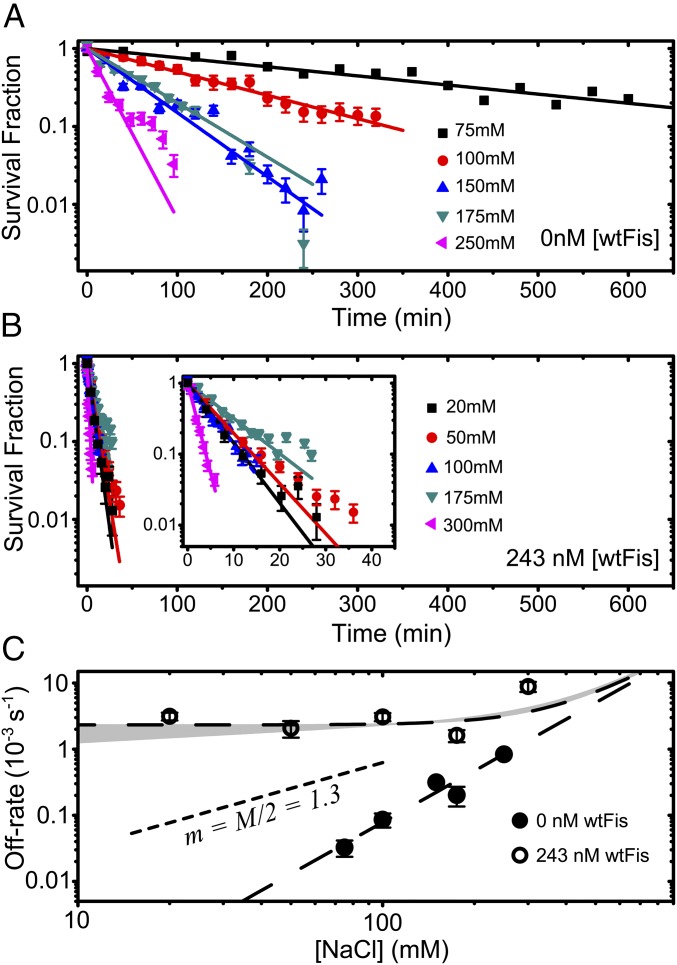
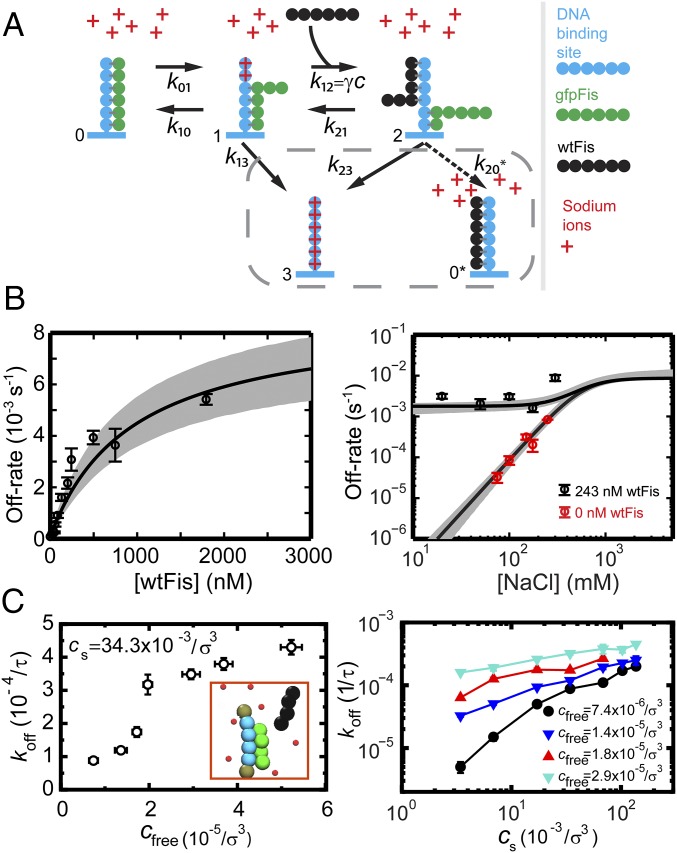
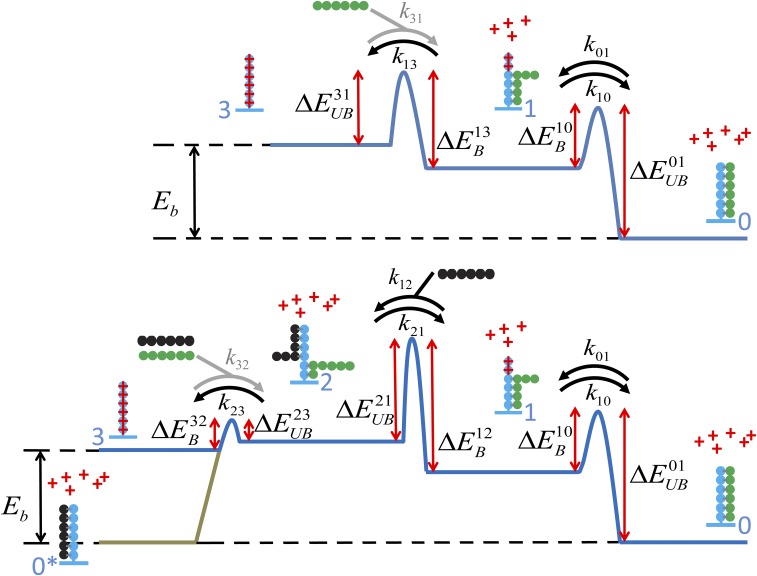
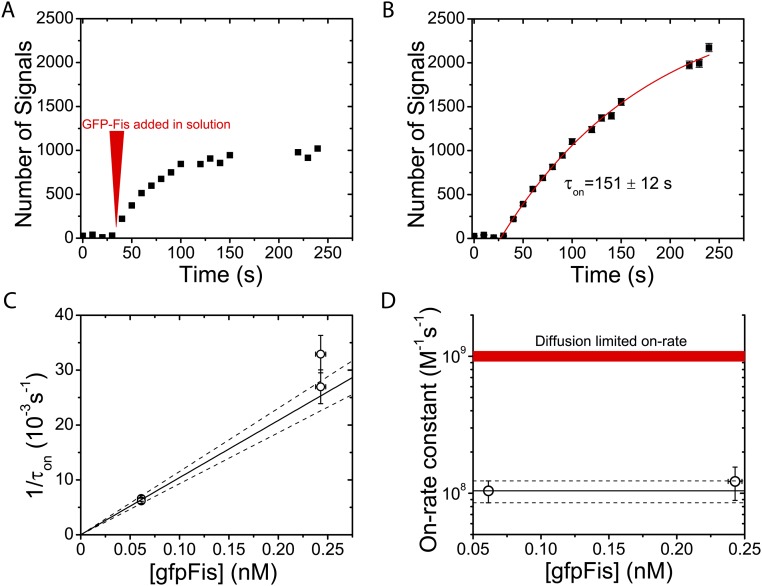
Similar articles
-
DNA-Segment-Facilitated Dissociation of Fis and NHP6A from DNA Detected via Single-Molecule Mechanical Response.J Mol Biol. 2015 Sep 25;427(19):3123-36. doi: 10.1016/j.jmb.2015.07.015. Epub 2015 Jul 26. J Mol Biol. 2015. PMID: 26220077 Free PMC article.
-
Facilitated Dissociation of a Nucleoid Protein from the Bacterial Chromosome.J Bacteriol. 2016 May 27;198(12):1735-42. doi: 10.1128/JB.00225-16. Print 2016 Jun 15. J Bacteriol. 2016. PMID: 27044624 Free PMC article.
-
Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA.Nucleic Acids Res. 2011 Mar;39(6):2249-59. doi: 10.1093/nar/gkq1140. Epub 2010 Nov 21. Nucleic Acids Res. 2011. PMID: 21097894 Free PMC article.
-
Nhp6: a small but powerful effector of chromatin structure in Saccharomyces cerevisiae.Biochim Biophys Acta. 2010 Jan-Feb;1799(1-2):175-80. doi: 10.1016/j.bbagrm.2009.11.010. Biochim Biophys Acta. 2010. PMID: 20123079 Free PMC article. Review.
-
The different (sur)faces of Rap1p.Mol Genet Genomics. 2003 Mar;268(6):791-8. doi: 10.1007/s00438-002-0801-3. Epub 2003 Jan 25. Mol Genet Genomics. 2003. PMID: 12655405 Review.
Cited by
-
DNA-facilitated target search by nucleoproteins: Extension of a biosensor-surface plasmon resonance method.Anal Biochem. 2021 Sep 15;629:114298. doi: 10.1016/j.ab.2021.114298. Epub 2021 Jul 10. Anal Biochem. 2021. PMID: 34252439 Free PMC article.
-
Intrinsically disordered domain of tumor suppressor p53 facilitates target search by ultrafast transfer between different DNA strands.Nucleic Acids Res. 2018 Aug 21;46(14):7261-7269. doi: 10.1093/nar/gky586. Nucleic Acids Res. 2018. PMID: 29986056 Free PMC article.
-
The HMGB chromatin protein Nhp6A can bypass obstacles when traveling on DNA.Nucleic Acids Res. 2020 Nov 4;48(19):10820-10831. doi: 10.1093/nar/gkaa799. Nucleic Acids Res. 2020. PMID: 32997109 Free PMC article.
-
Single-molecule imaging correlates decreasing nuclear volume with increasing TF-chromatin associations during zebrafish development.Nat Commun. 2018 Dec 6;9(1):5218. doi: 10.1038/s41467-018-07731-8. Nat Commun. 2018. PMID: 30523256 Free PMC article.
-
An in-silico human cell model reveals the influence of spatial organization on RNA splicing.PLoS Comput Biol. 2020 Mar 25;16(3):e1007717. doi: 10.1371/journal.pcbi.1007717. eCollection 2020 Mar. PLoS Comput Biol. 2020. PMID: 32210422 Free PMC article.
References
-
- Dominguez-Sola D, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–451. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
