

Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis
- PMID: 28366453
- PMCID: PMC5390134
- DOI: 10.1016/j.stemcr.2017.02.019
Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis
Abstract
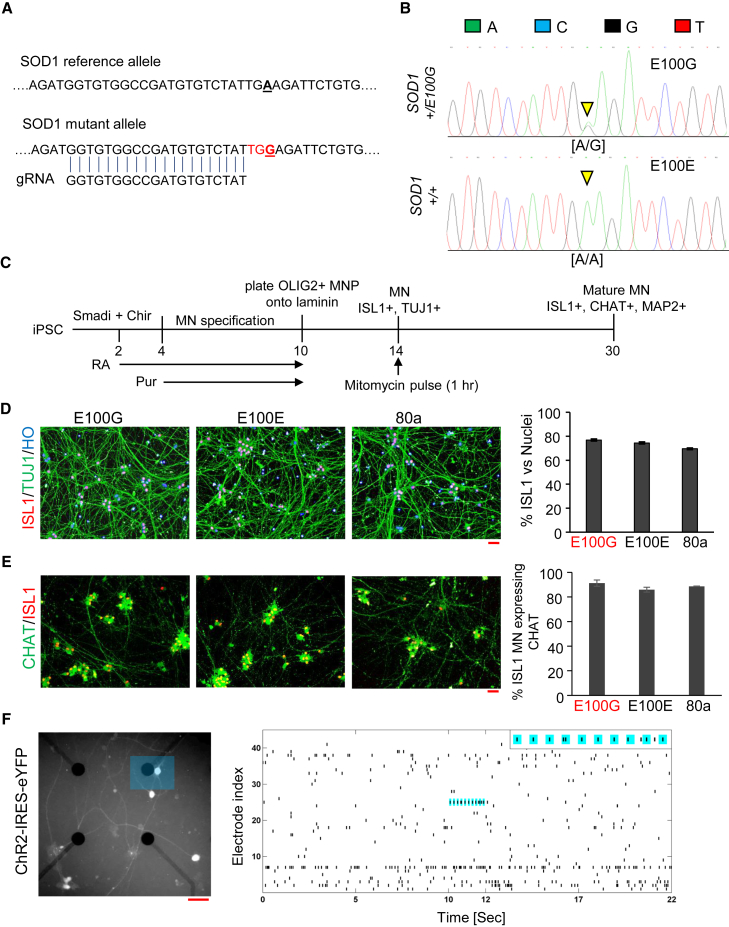
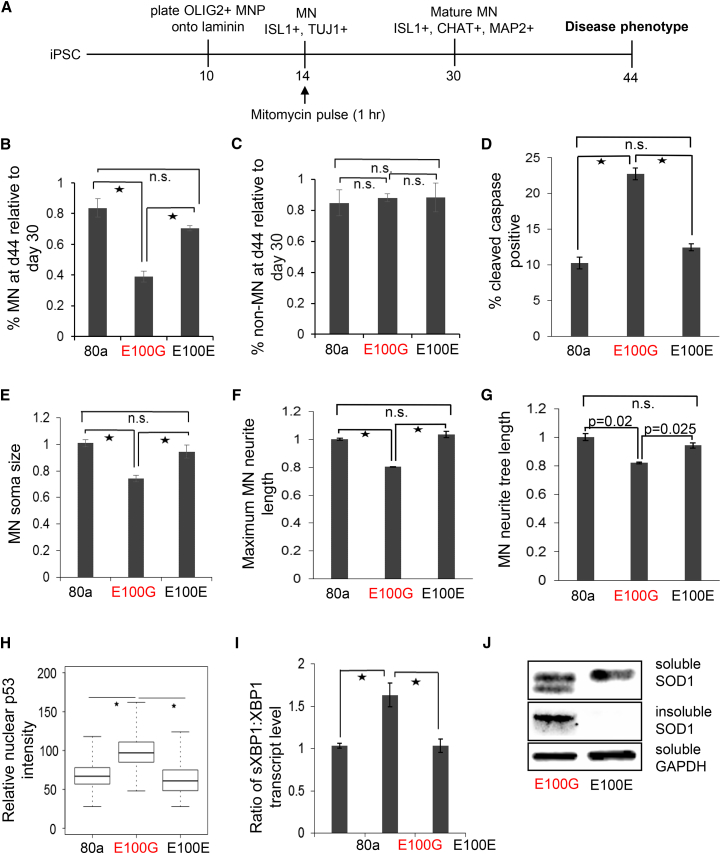
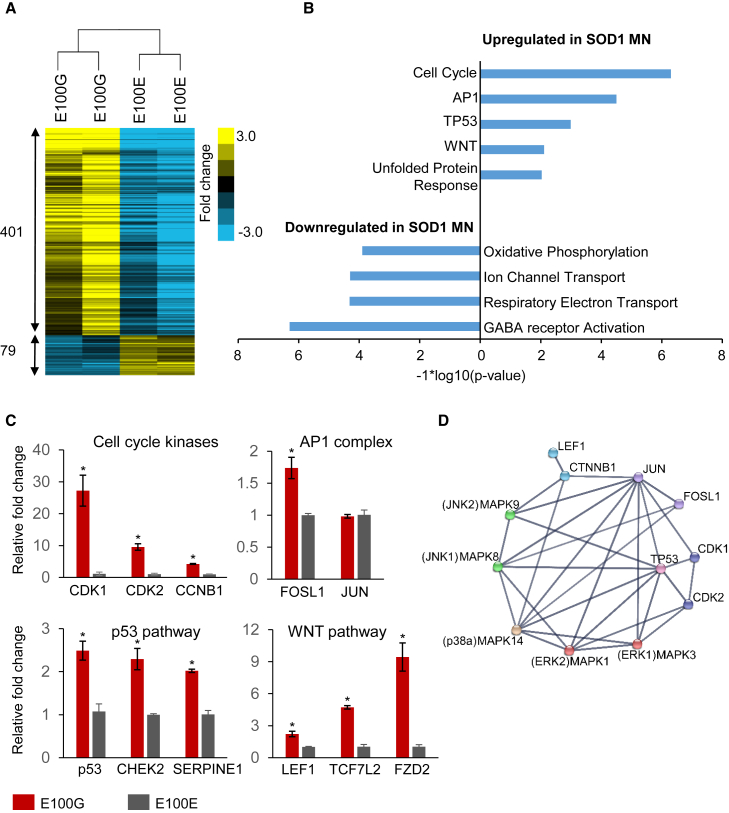
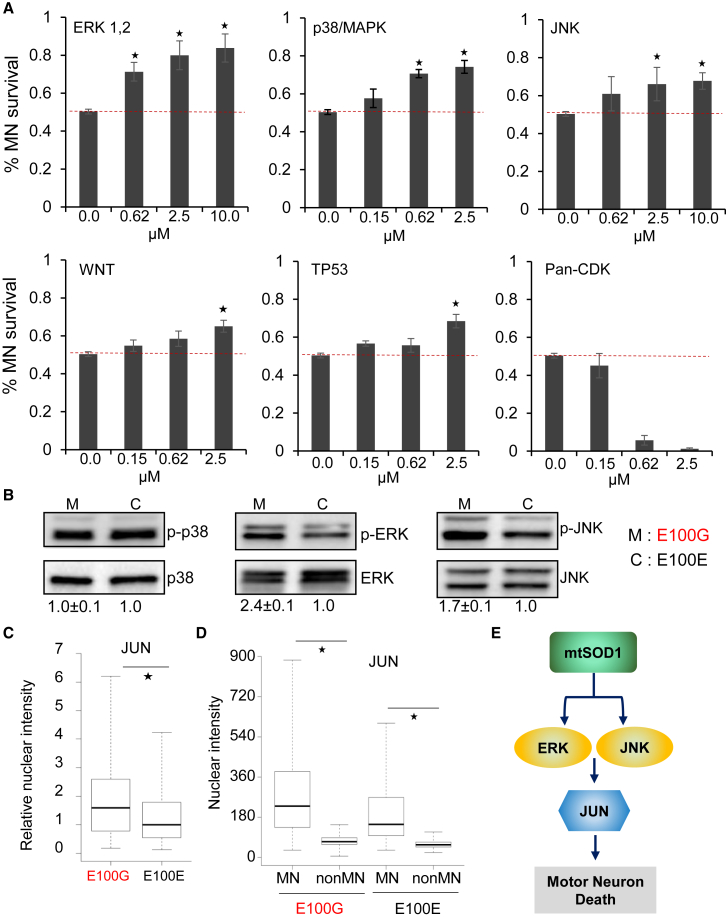
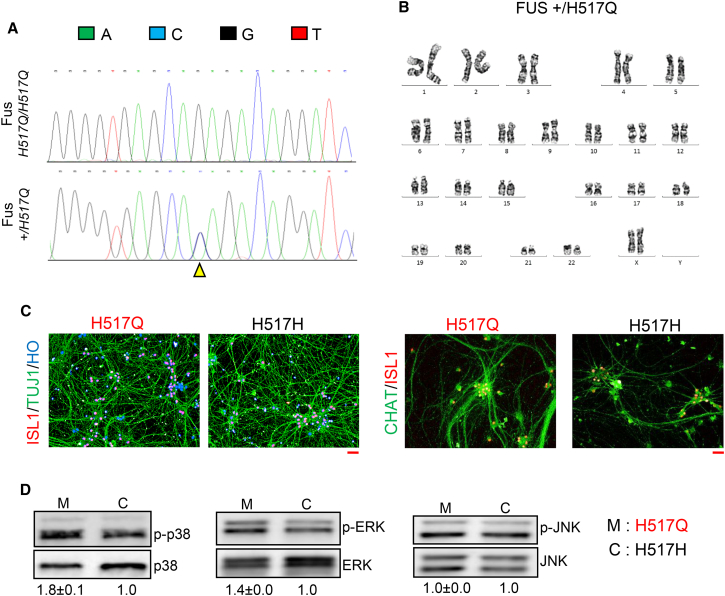
Although mutations in several genes with diverse functions have been known to cause amyotrophic lateral sclerosis (ALS), it is unknown to what extent causal mutations impinge on common pathways that drive motor neuron (MN)-specific neurodegeneration. In this study, we combined induced pluripotent stem cells-based disease modeling with genome engineering and deep RNA sequencing to identify pathways dysregulated by mutant SOD1 in human MNs. Gene expression profiling and pathway analysis followed by pharmacological screening identified activated ERK and JNK signaling as key drivers of neurodegeneration in mutant SOD1 MNs. The AP1 complex member JUN, an ERK/JNK downstream target, was observed to be highly expressed in MNs compared with non-MNs, providing a mechanistic insight into the specific degeneration of MNs. Importantly, investigations of mutant FUS MNs identified activated p38 and ERK, indicating that network perturbations induced by ALS-causing mutations converge partly on a few specific pathways that are drug responsive and provide immense therapeutic potential.
Keywords: ALS; CRISPR-Cas9; ERK; FUS; JNK; JUN; SOD1; TP53; WNT; p38.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Al-Chalabi A., Jones A., Troakes C., King A., Al-Sarraj S., van den Berg L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta. Neuropathol. 2012;124:339–352. - PubMed
-
- Andersen P.M., Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 2011;7:603–615. - PubMed
-
- Bilican B., Serio A., Barmada S.J., Nishimura A.L., Sullivan G.J., Carrasco M., Phatnani H.P., Puddifoot C.A., Story D., Fletcher J. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA. 2012;109:5803–5808. - PMC - PubMed
-
- Dafinca R., Scaber J., Ababneh N., Lalic T., Weir G., Christian H., Vowles J., Douglas A.G., Fletcher-Jones A., Browne C. C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells. 2016;34:2063–2078. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
