

SLIMM: species level identification of microorganisms from metagenomes
- PMID: 28367376
- PMCID: PMC5372838
- DOI: 10.7717/peerj.3138
SLIMM: species level identification of microorganisms from metagenomes
Abstract
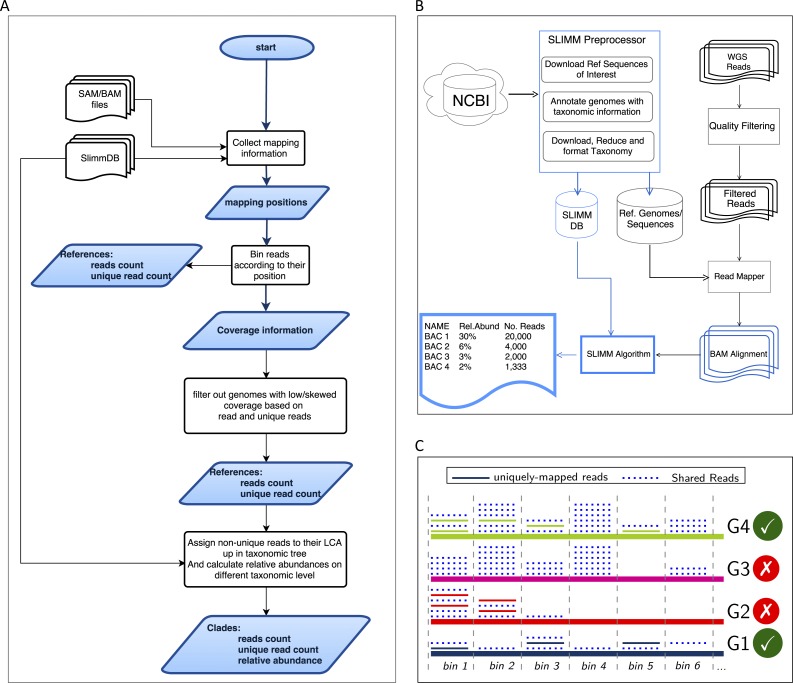
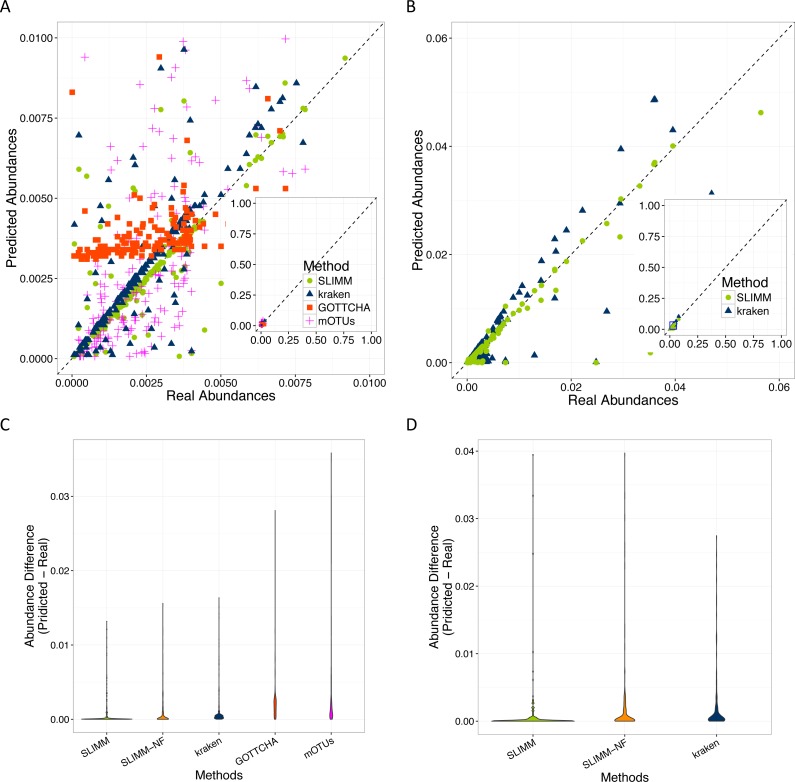
Identification and quantification of microorganisms is a significant step in studying the alpha and beta diversities within and between microbial communities respectively. Both identification and quantification of a given microbial community can be carried out using whole genome shotgun sequences with less bias than when using 16S-rDNA sequences. However, shared regions of DNA among reference genomes and taxonomic units pose a significant challenge in assigning reads correctly to their true origins. The existing microbial community profiling tools commonly deal with this problem by either preparing signature-based unique references or assigning an ambiguous read to its least common ancestor in a taxonomic tree. The former method is limited to making use of the reads which can be mapped to the curated regions, while the latter suffer from the lack of uniquely mapped reads at lower (more specific) taxonomic ranks. Moreover, even if the tools exhibited good performance in calling the organisms present in a sample, there is still room for improvement in determining the correct relative abundance of the organisms. We present a new method Species Level Identification of Microorganisms from Metagenomes (SLIMM) which addresses the above issues by using coverage information of reference genomes to remove unlikely genomes from the analysis and subsequently gain more uniquely mapped reads to assign at lower ranks of a taxonomic tree. SLIMM is based on a few, seemingly easy steps which when combined create a tool that outperforms state-of-the-art tools in run-time and memory usage while being on par or better in computing quantitative and qualitative information at species-level.
Keywords: Metagenomics; Microbial communities; Microbiology; Microorganisms; NGS data; Taxonomic profiling.
Conflict of interest statement
The authors declare there are no competing interests.
Figures

References
-
- Brown CT, Howe A, Zhang Q, Pyrkosz AB, Brom TH. A reference-free algorithm for computational normalization of shotgun sequencing data. 20121203.4802
-
- Francis OE, Bendall M, Manimaran S, Hong C, Clement NL, Castro-Nallar E, Snell Q, Schaalje GB, Clement MJ, Crandall KA, Johnson WE. Pathoscope: species identification and strain attribution with unassembled sequencing data. Genome Research. 2013;23(10):1721–1729. doi: 10.1101/gr.150151.112. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
