

The conserved protein Seb1 drives transcription termination by binding RNA polymerase II and nascent RNA
- PMID: 28367989
- PMCID: PMC5382271
- DOI: 10.1038/ncomms14861
The conserved protein Seb1 drives transcription termination by binding RNA polymerase II and nascent RNA
Abstract
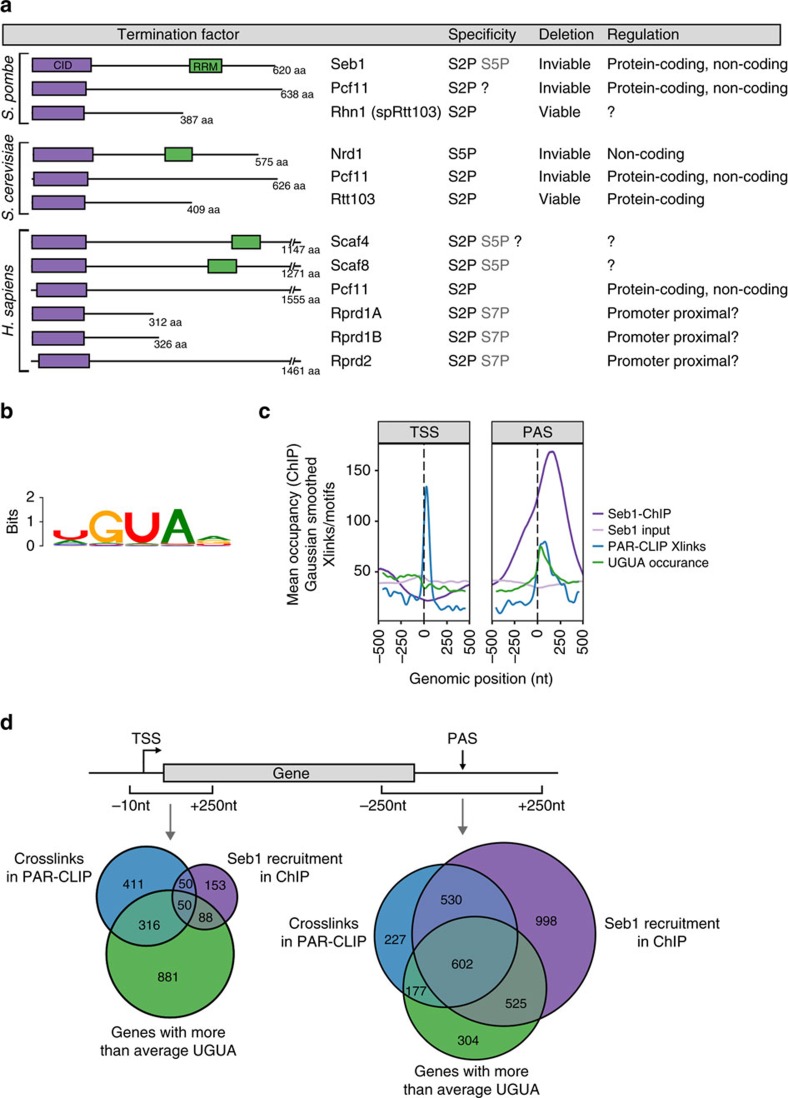
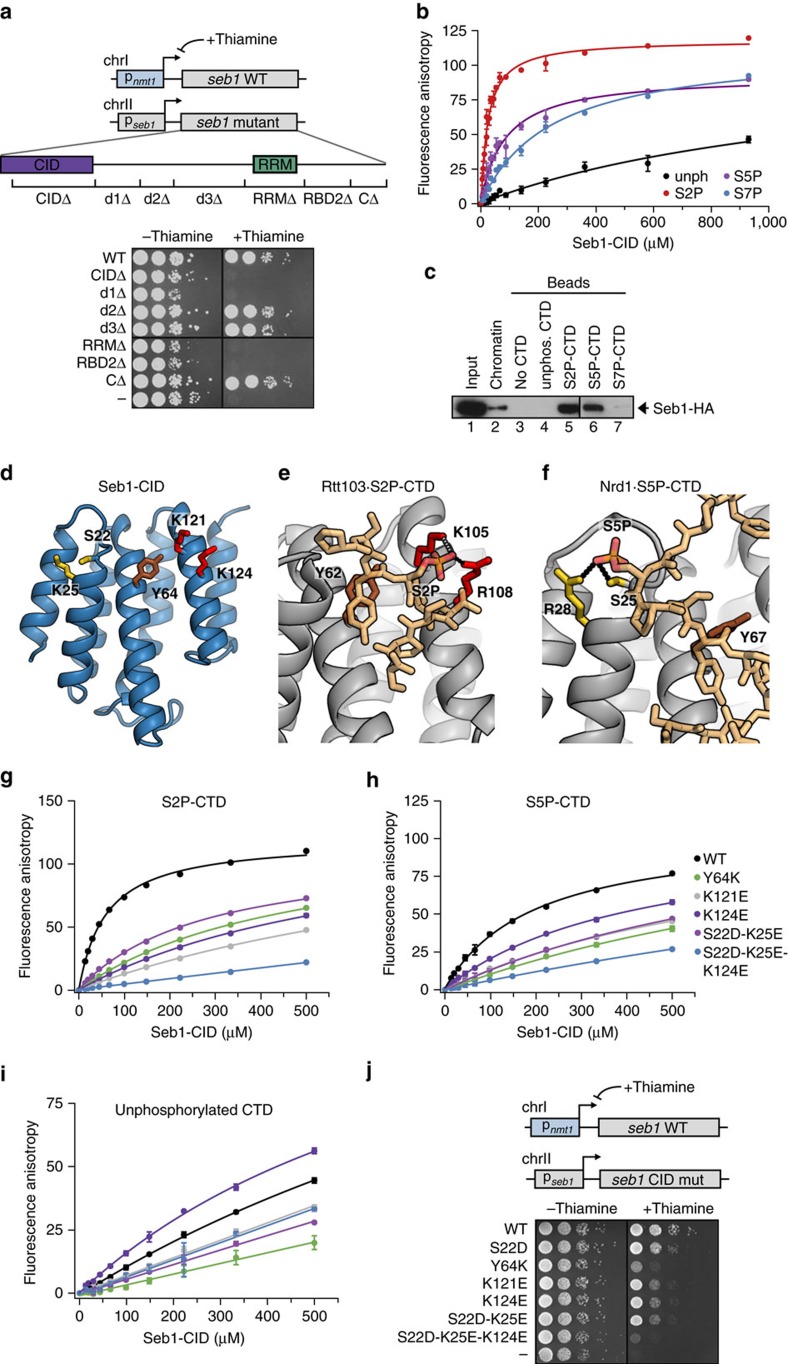
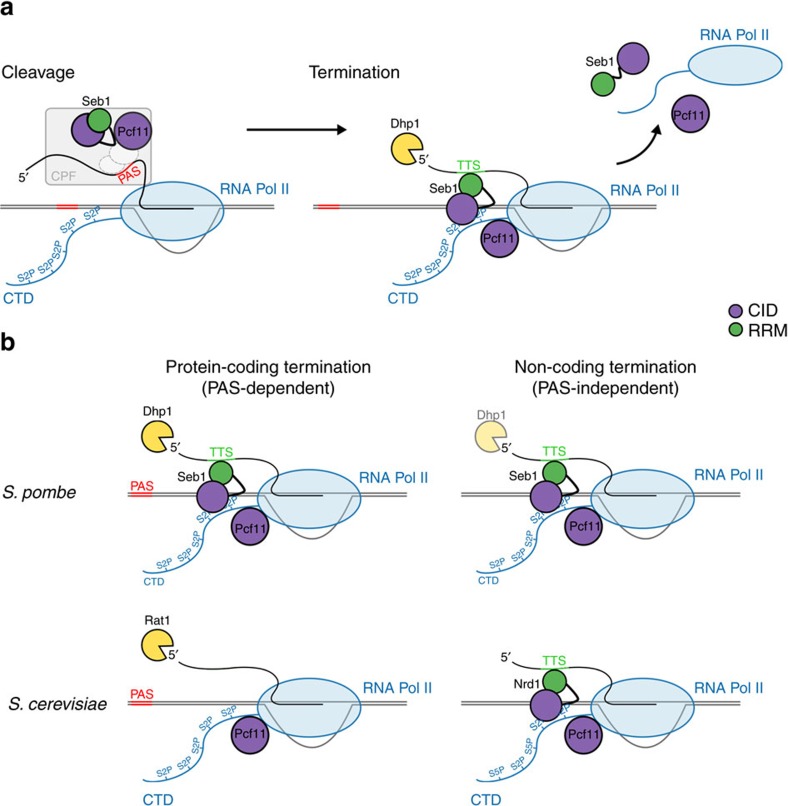
Termination of RNA polymerase II (Pol II) transcription is an important step in the transcription cycle, which involves the dislodgement of polymerase from DNA, leading to release of a functional transcript. Recent studies have identified the key players required for this process and showed that a common feature of these proteins is a conserved domain that interacts with the phosphorylated C-terminus of Pol II (CTD-interacting domain, CID). However, the mechanism by which transcription termination is achieved is not understood. Using genome-wide methods, here we show that the fission yeast CID-protein Seb1 is essential for termination of protein-coding and non-coding genes through interaction with S2-phosphorylated Pol II and nascent RNA. Furthermore, we present the crystal structures of the Seb1 CTD- and RNA-binding modules. Unexpectedly, the latter reveals an intertwined two-domain arrangement of a canonical RRM and second domain. These results provide important insights into the mechanism underlying eukaryotic transcription termination.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
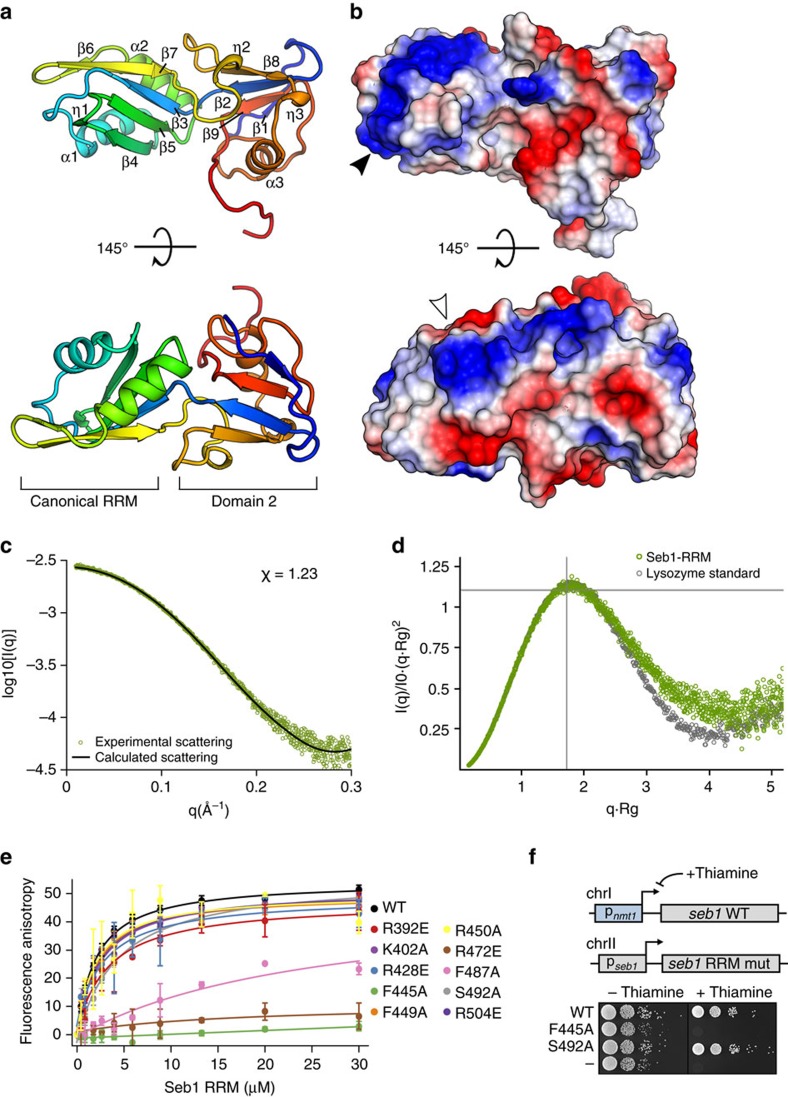
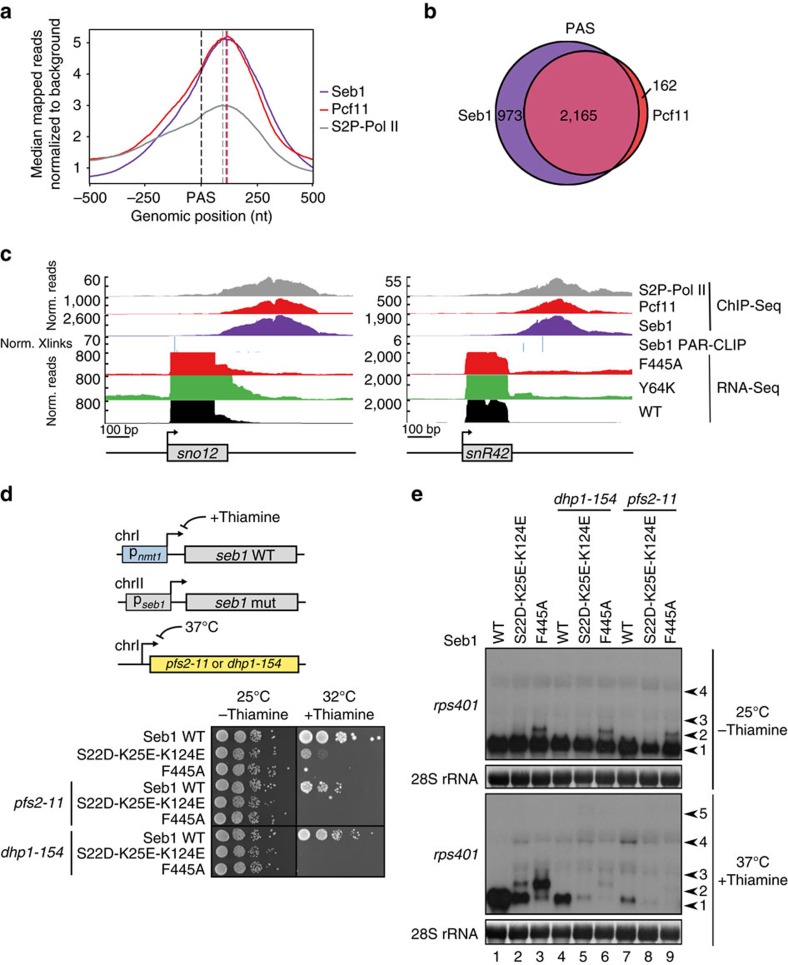
 , 1.104), the peak position of an ideal globular and rigid protein. Rigid proteins show a characteristic parabolic shape with a peak at the indicated position (as is the case here), while unfolded proteins would plateau with increasing q-values. (e) Analysis of Seb1-SUMO-RRM388–540 binding to FAM-tagged AUUAGUAAAA RNA by FA. Error bars indicate standard deviation of three technical replicates. (f) Spot test showing the effect of the indicated Seb1-RRM point mutations on cell growth.
, 1.104), the peak position of an ideal globular and rigid protein. Rigid proteins show a characteristic parabolic shape with a peak at the indicated position (as is the case here), while unfolded proteins would plateau with increasing q-values. (e) Analysis of Seb1-SUMO-RRM388–540 binding to FAM-tagged AUUAGUAAAA RNA by FA. Error bars indicate standard deviation of three technical replicates. (f) Spot test showing the effect of the indicated Seb1-RRM point mutations on cell growth.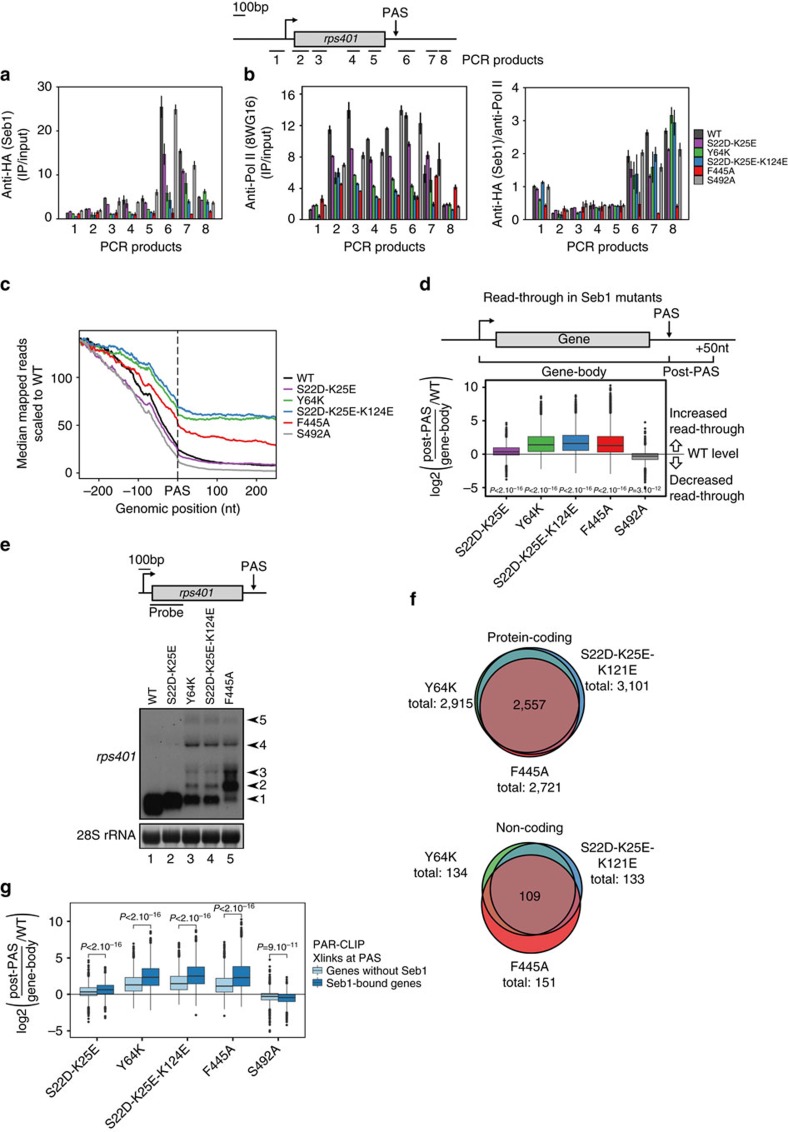
References
-
- Martens J. A., Laprade L. & Winston F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 429, 571–574 (2004). - PubMed
-
- Kim M. et al. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432, 517–522 (2004). - PubMed
-
- Kobor M. S. & Greenblatt J. Regulation of transcription elongation by phosphorylation. Biochim. Biophys. Acta 1577, 261–275 (2002). - PubMed
-
- Buratowski S. The CTD code. Nat. Struct. Biol. 10, 679–680 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
