

Estimating the selective effects of heterozygous protein-truncating variants from human exome data
- PMID: 28369035
- PMCID: PMC5618255
- DOI: 10.1038/ng.3831
Estimating the selective effects of heterozygous protein-truncating variants from human exome data
Abstract
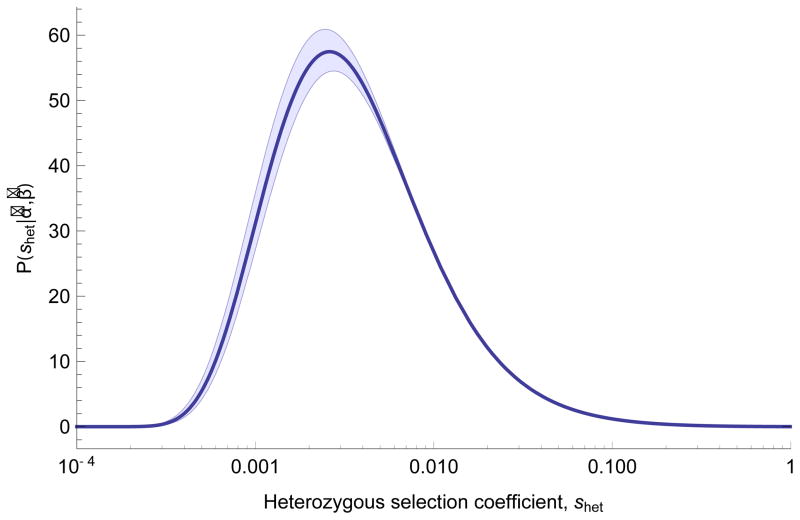
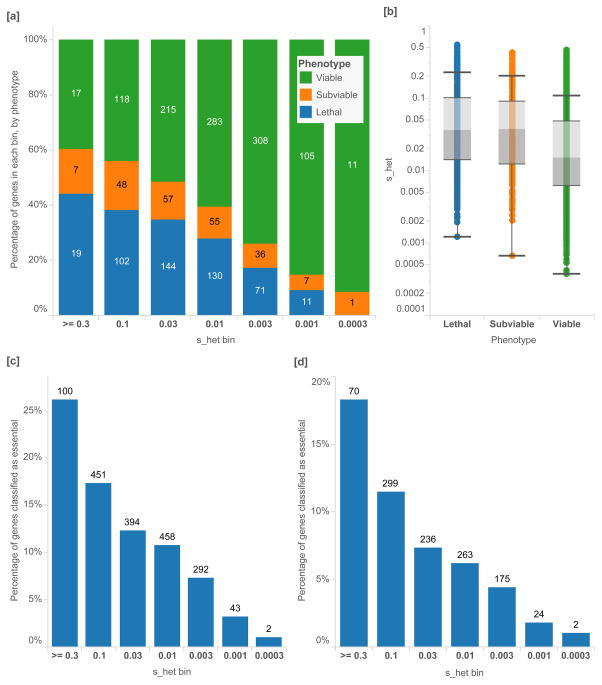
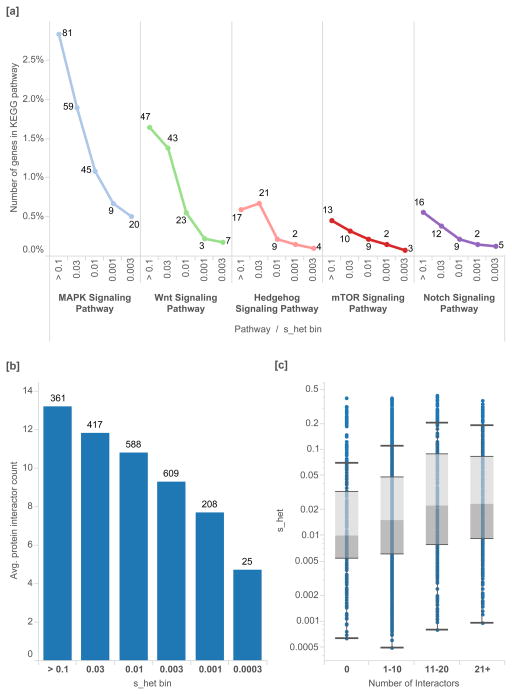
The evolutionary cost of gene loss is a central question in genetics and has been investigated in model organisms and human cell lines. In humans, tolerance of the loss of one or both functional copies of a gene is related to the gene's causal role in disease. However, estimates of the selection and dominance coefficients in humans have been elusive. Here we analyze exome sequence data from 60,706 individuals to make genome-wide estimates of selection against heterozygous loss of gene function. Using this distribution of selection coefficients for heterozygous protein-truncating variants (PTVs), we provide corresponding Bayesian estimates for individual genes. We find that genes under the strongest selection are enriched in embryonic lethal mouse knockouts, Mendelian disease-associated genes, and regulators of transcription. Screening by essentiality, we find a large set of genes under strong selection that are likely to have crucial functions but have not yet been thoroughly characterized.
Conflict of interest statement
The authors have no competing interests as defined by Springer Nature, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Figures
Comment in
-
Selective effects of heterozygous protein-truncating variants.Nat Genet. 2019 Jan;51(1):2. doi: 10.1038/s41588-018-0291-9. Nat Genet. 2019. PMID: 30478439 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
