

The impact of structural variation on human gene expression
- PMID: 28369037
- PMCID: PMC5406250
- DOI: 10.1038/ng.3834
The impact of structural variation on human gene expression
Abstract
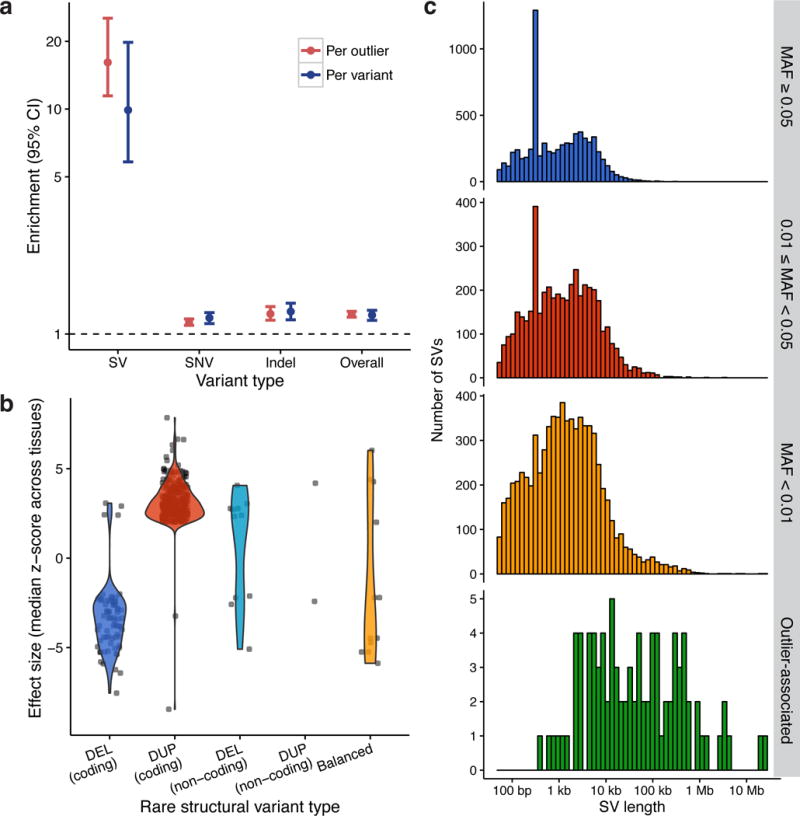
Structural variants (SVs) are an important source of human genetic diversity, but their contribution to traits, disease and gene regulation remains unclear. We mapped cis expression quantitative trait loci (eQTLs) in 13 tissues via joint analysis of SVs, single-nucleotide variants (SNVs) and short insertion/deletion (indel) variants from deep whole-genome sequencing (WGS). We estimated that SVs are causal at 3.5-6.8% of eQTLs-a substantially higher fraction than prior estimates-and that expression-altering SVs have larger effect sizes than do SNVs and indels. We identified 789 putative causal SVs predicted to directly alter gene expression: most (88.3%) were noncoding variants enriched at enhancers and other regulatory elements, and 52 were linked to genome-wide association study loci. We observed a notable abundance of rare high-impact SVs associated with aberrant expression of nearby genes. These results suggest that comprehensive WGS-based SV analyses will increase the power of common- and rare-variant association studies.
Conflict of interest statement
D.F.C. is a paid consultant of PierianDx. The authors declare no other competing financial interests.
Figures
References
MeSH terms
Grants and funding
- R01 DA006227/DA/NIDA NIH HHS/United States
- R01 MH101782/MH/NIMH NIH HHS/United States
- R01 MH101810/MH/NIMH NIH HHS/United States
- R01 MH101819/MH/NIMH NIH HHS/United States
- R01 DA033684/DA/NIDA NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- UM1 HG008853/HG/NHGRI NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- T32 HG000044/HG/NHGRI NIH HHS/United States
- R01 MH101822/MH/NIMH NIH HHS/United States
- R01 HG007178/HG/NHGRI NIH HHS/United States
- R01 MH101820/MH/NIMH NIH HHS/United States
- R01 MH101825/MH/NIMH NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- K22 HG000044/HG/NHGRI NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
