

A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy
- PMID: 28369220
- PMCID: PMC6248622
- DOI: 10.1093/brain/awx058
A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy
Abstract
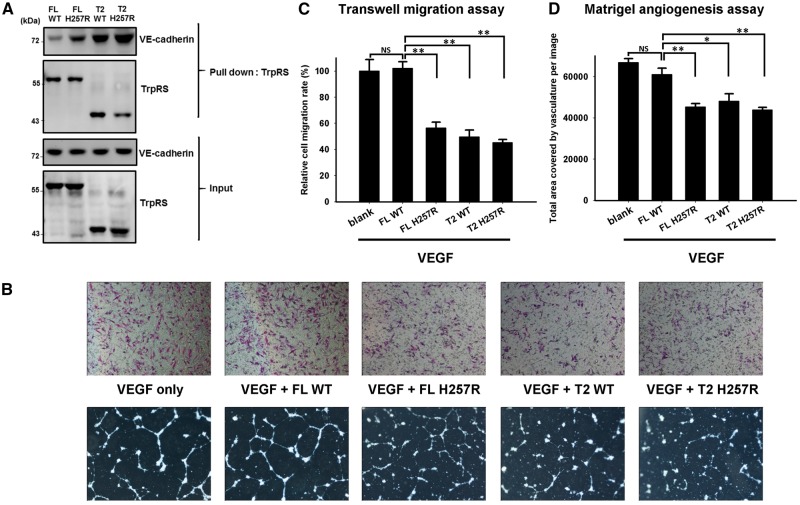
Distal hereditary motor neuropathy is a heterogeneous group of inherited neuropathies characterized by distal limb muscle weakness and atrophy. Although at least 15 genes have been implicated in distal hereditary motor neuropathy, the genetic causes remain elusive in many families. To identify an additional causal gene for distal hereditary motor neuropathy, we performed exome sequencing for two affected individuals and two unaffected members in a Taiwanese family with an autosomal dominant distal hereditary motor neuropathy in which mutations in common distal hereditary motor neuropathy-implicated genes had been excluded. The exome sequencing revealed a heterozygous mutation, c.770A > G (p.His257Arg), in the cytoplasmic tryptophanyl-tRNA synthetase (TrpRS) gene (WARS) that co-segregates with the neuropathy in the family. Further analyses of WARS in an additional 79 Taiwanese pedigrees with inherited neuropathies and 163 index cases from Australian, European, and Korean distal hereditary motor neuropathy families identified the same mutation in another Taiwanese distal hereditary motor neuropathy pedigree with different ancestries and one additional Belgian distal hereditary motor neuropathy family of Caucasian origin. Cell transfection studies demonstrated a dominant-negative effect of the p.His257Arg mutation on aminoacylation activity of TrpRS, which subsequently compromised protein synthesis and reduced cell viability. His257Arg TrpRS also inhibited neurite outgrowth and led to neurite degeneration in the neuronal cell lines and rat motor neurons. Further in vitro analyses showed that the WARS mutation could potentiate the angiostatic activities of TrpRS by enhancing its interaction with vascular endothelial-cadherin. Taken together, these findings establish WARS as a gene whose mutations may cause distal hereditary motor neuropathy and alter canonical and non-canonical functions of TrpRS.
Keywords: WARS; dHMN; distal hereditary motor neuropathy; exome sequencing; tryptophanyl-tRNA synthetase.
© The Author (2017). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
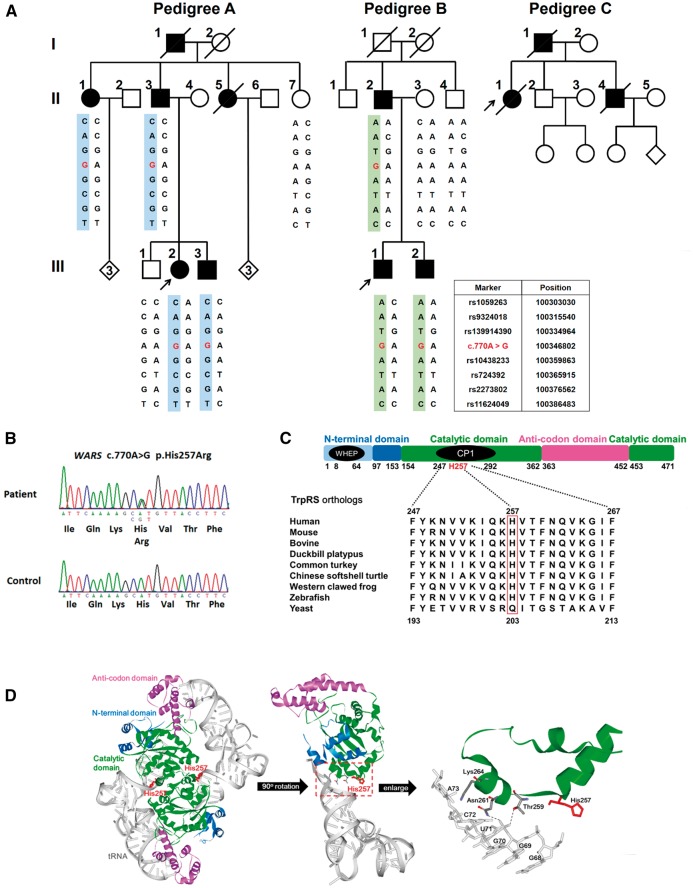
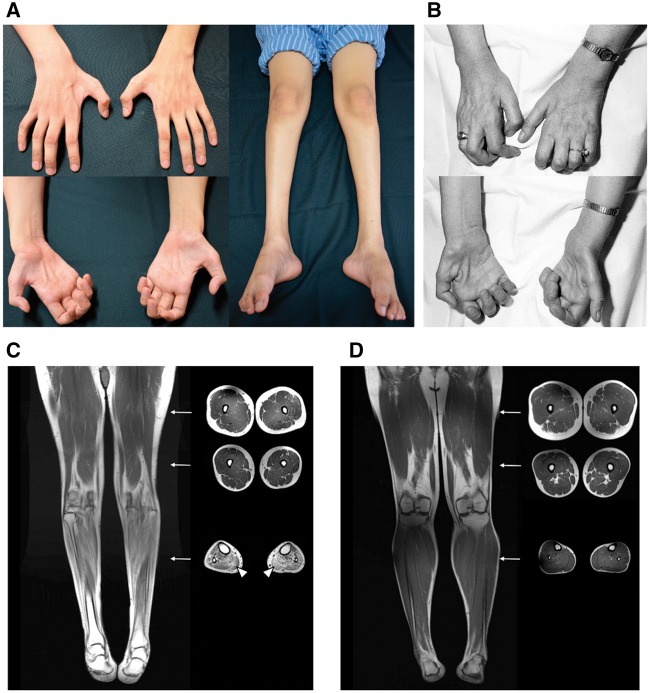
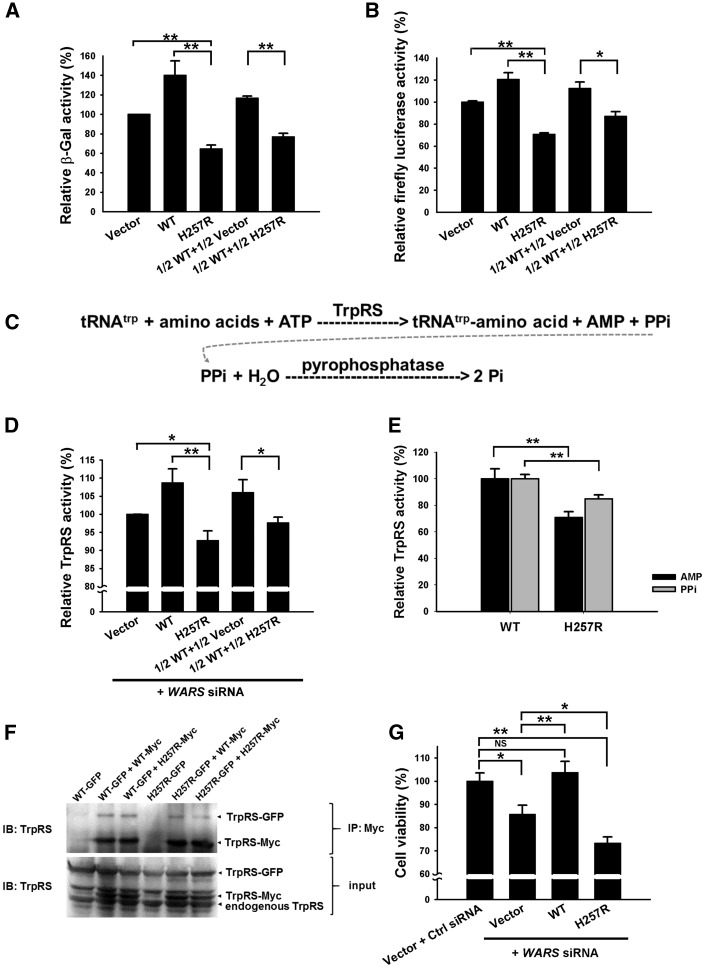
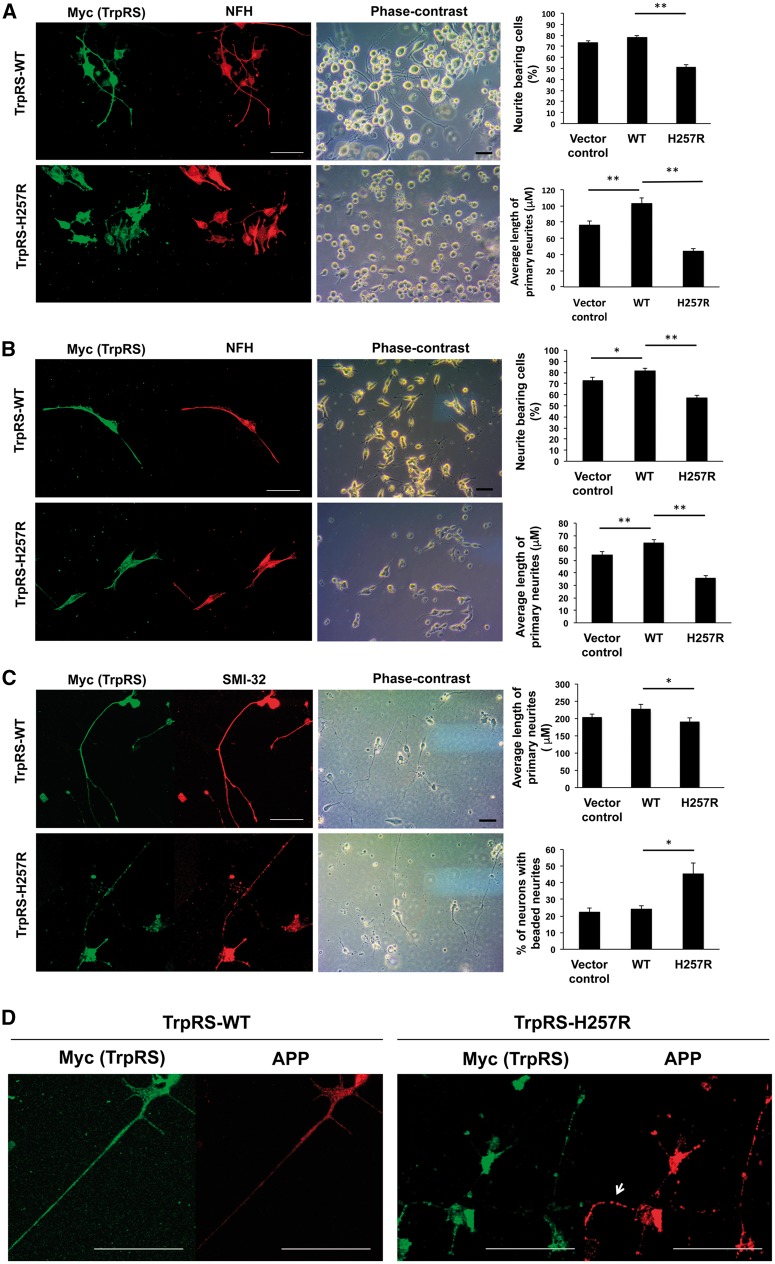
Comment in
-
A novel WARS mutation causes distal hereditary motor neuropathy in a Chinese family.Brain. 2019 Sep 1;142(9):e49. doi: 10.1093/brain/awz218. Brain. 2019. PMID: 31321409 No abstract available.
References
-
- Delarue M. Aminoacyl-tRNA synthestases. Curr Opin Struct Biol 1995; 5: 48–55. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
