

The Influence of HIV on the Evolution of Mycobacterium tuberculosis
- PMID: 28369607
- PMCID: PMC5455964
- DOI: 10.1093/molbev/msx107
The Influence of HIV on the Evolution of Mycobacterium tuberculosis
Abstract
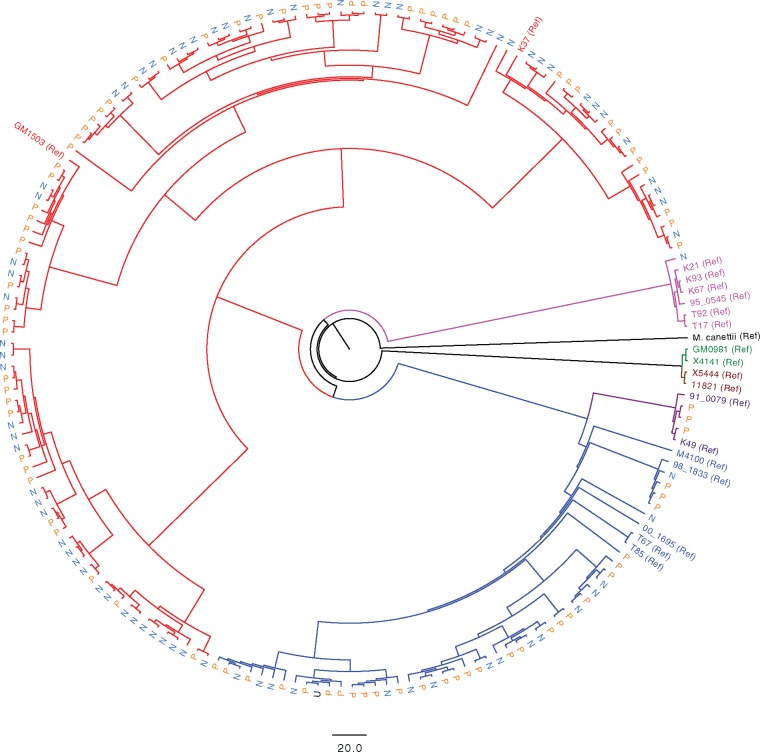
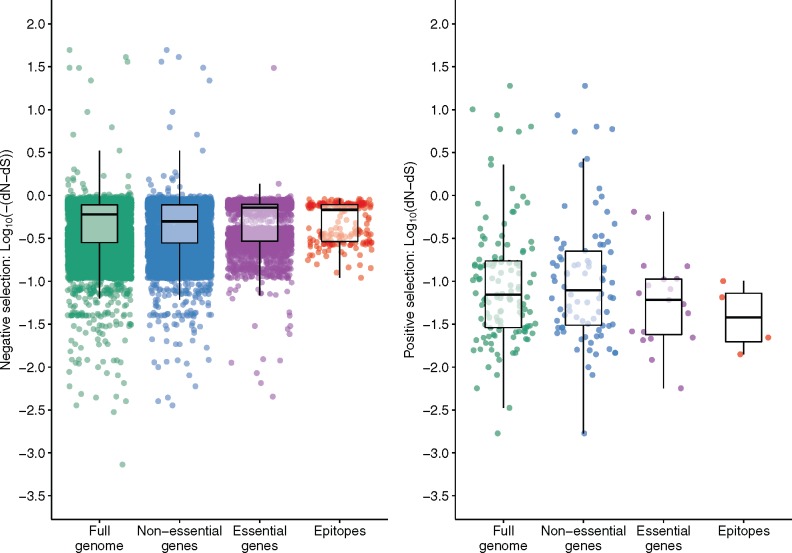
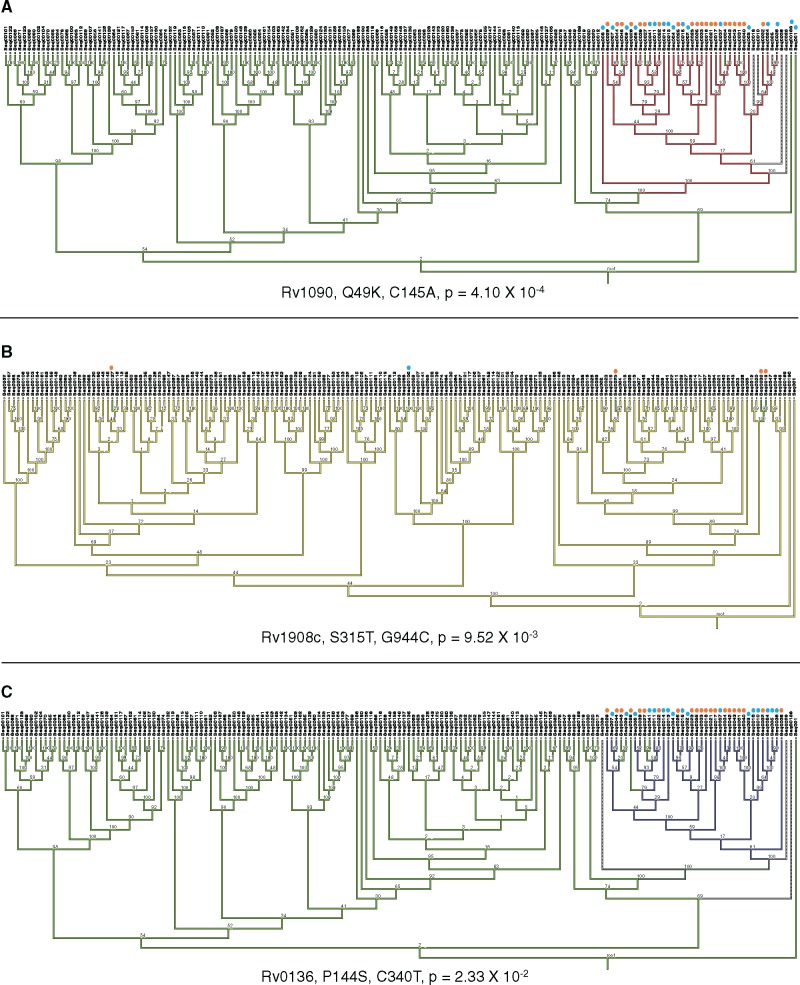
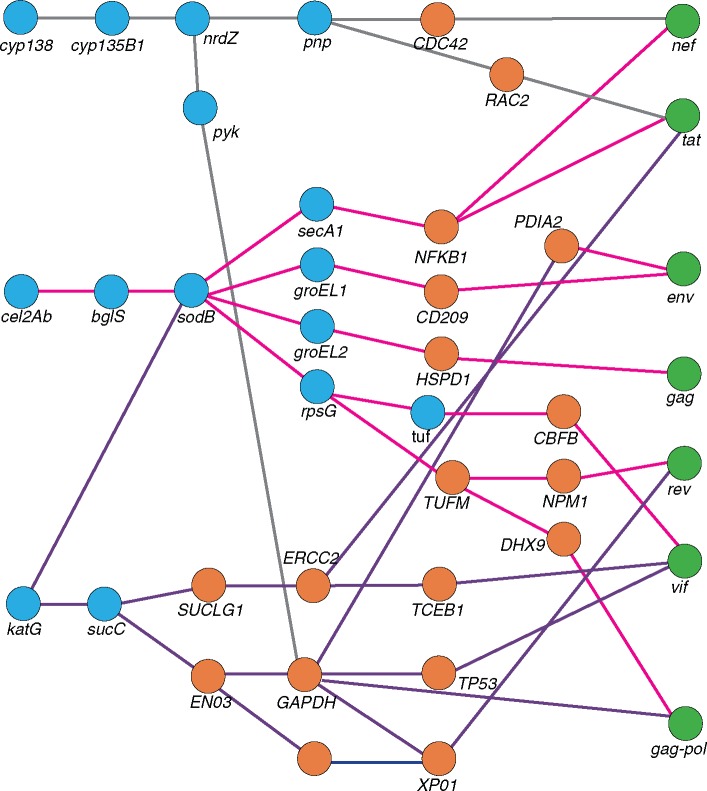
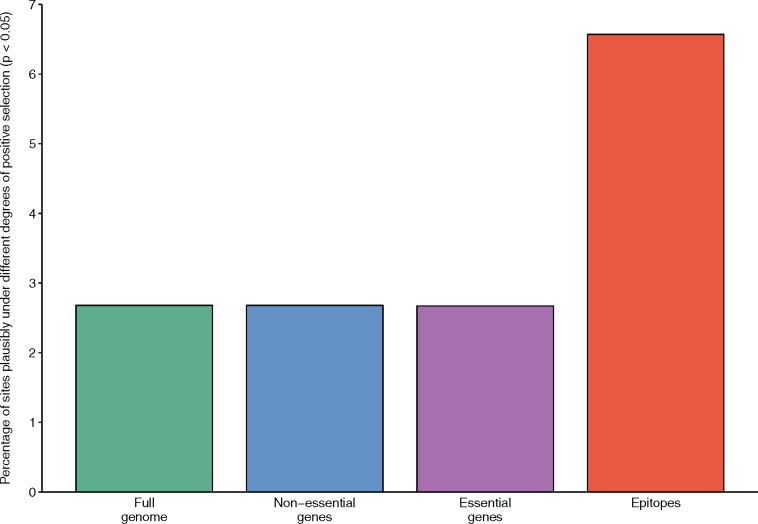
HIV significantly affects the immunological environment during tuberculosis coinfection, and therefore may influence the selective landscape upon which M. tuberculosis evolves. To test this hypothesis whole genome sequences were determined for 169 South African M. tuberculosis strains from HIV-1 coinfected and uninfected individuals and analyzed using two Bayesian codon-model based selection analysis approaches: FUBAR which was used to detect persistent positive and negative selection (selection respectively favoring and disfavoring nonsynonymous substitutions); and MEDS which was used to detect episodic directional selection specifically favoring nonsynonymous substitutions within HIV-1 infected individuals. Among the 25,251 polymorphic codon sites analyzed, FUBAR revealed that 189-fold more were detectably evolving under persistent negative selection than were evolving under persistent positive selection. Three specific codon sites within the genes celA2b, katG, and cyp138 were identified by MEDS as displaying significant evidence of evolving under directional selection influenced by HIV-1 coinfection. All three genes encode proteins that may indirectly interact with human proteins that, in turn, interact functionally with HIV proteins. Unexpectedly, epitope encoding regions were enriched for sites displaying weak evidence of directional selection influenced by HIV-1. Although the low degree of genetic diversity observed in our M. tuberculosis data set means that these results should be interpreted carefully, the effects of HIV-1 on epitope evolution in M. tuberculosis may have implications for the design of M. tuberculosis vaccines that are intended for use in populations with high HIV-1 infection rates.
Keywords: HIV coinfection; Mycobacterium tuberculosis; evolution; natural selection.
© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Abraha A, Nankya IL, Gibson R, Demers K, Tebit DM, Johnston E, Katzenstein D, Siddiqui A, Herrera C, Fischetti L, et al.2009. CCR5- and CXCR4-tropic subtype C human immunodeficiency virus type 1 isolates have a lower level of pathogenic fitness than other dominant group M subtypes: implications for the epidemic. J Virol. 83:5592–5605. - PMC - PubMed
-
- Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol. 62:53–70. - PubMed
-
- Anisimova M, Kosiol C.. 2009. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 26:255–271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 203135/WT_/Wellcome Trust/United Kingdom
- FC00110218/CRUK_/Cancer Research UK/United Kingdom
- 104803/WT_/Wellcome Trust/United Kingdom
- U19 AI111276/AI/NIAID NIH HHS/United States
- FC00110218/MRC_/Medical Research Council/United Kingdom
- MC_U117588499/MRC_/Medical Research Council/United Kingdom
- U01 AI115940/AI/NIAID NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- 309540/ERC_/European Research Council/International
- U01 AI069924/AI/NIAID NIH HHS/United States
- FC00110218/WT_/Wellcome Trust/United Kingdom
- 098051/WT_/Wellcome Trust/United Kingdom
