

Identification and characterization of a novel DGAT1 missense mutation associated with congenital diarrhea
- PMID: 28373485
- PMCID: PMC5454518
- DOI: 10.1194/jlr.P075119
Identification and characterization of a novel DGAT1 missense mutation associated with congenital diarrhea
Abstract
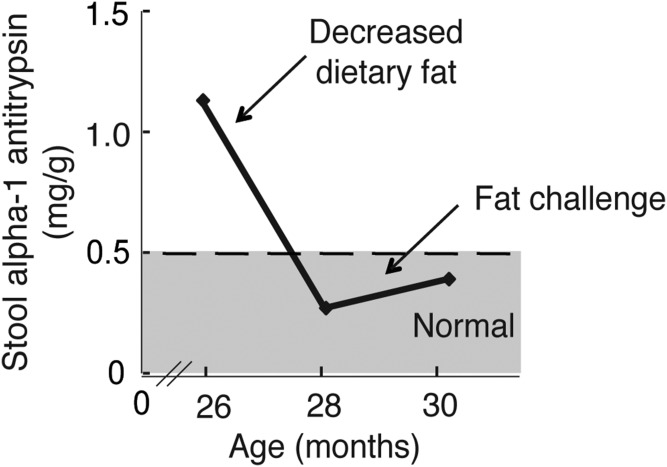
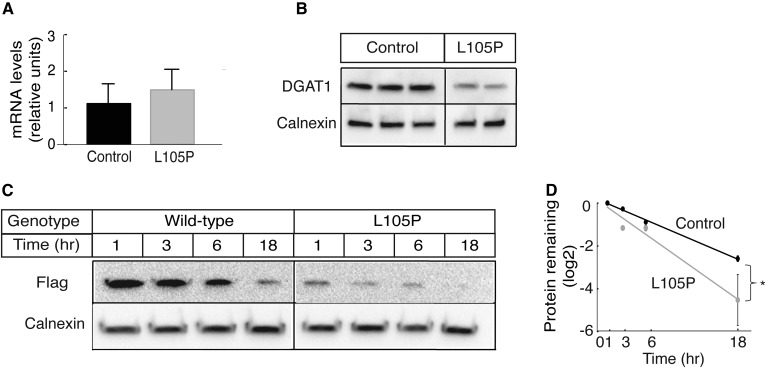
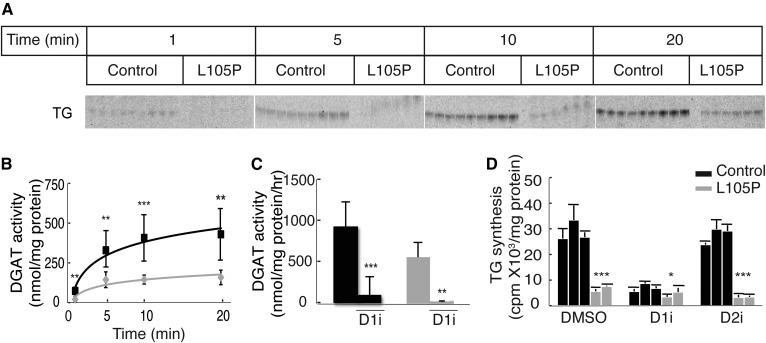
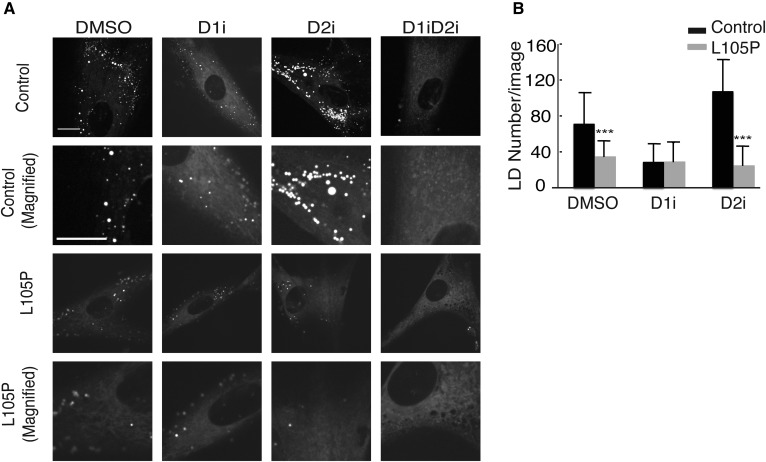
Acyl-CoA:diacylglycerol acyltransferase (DGAT)1 and DGAT2 catalyze triglyceride (TG) biosynthesis in humans. Biallelic loss-of-function mutations in human DGAT1 result in severe congenital diarrhea and protein-losing enteropathy. Additionally, pharmacologic inhibition of DGAT1 led to dose-related diarrhea in human clinical trials. Here we identify a previously unknown DGAT1 mutation in identical twins of South Asian descent. These male patients developed watery diarrhea shortly after birth, with protein-losing enteropathy and failure to thrive. Exome sequencing revealed a homozygous recessive mutation in DGAT1, c.314T>C, p.L105P. We show here that the p.L105P DGAT1 enzyme produced from the mutant allele is less abundant, resulting in partial loss of TG synthesis activity and decreased formation of lipid droplets in patient-derived primary dermal fibroblasts. Thus, in contrast with complete loss-of-function alleles of DGAT1, the p.L105P missense allele partially reduces TG synthesis activity and causes a less severe clinical phenotype. Our findings add to the growing recognition of DGAT1 deficiency as a cause of congenital diarrhea with protein-losing enteropathy and indicate that DGAT1 mutations result in a spectrum of diseases.
Keywords: acyl CoA:diacylglycerol acyltransferase; diet and dietary lipids; genetics; intestine; lipid droplets; protein-losing enteropathy; triglycerides.
Copyright © 2017 by the American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
Tobias Walther is an Investigator of the Howard Hughes Medical Institute.
Figures
References
-
- Cases S., Smith S. J., Zheng Y-W., Myers H. M., Lear S. R., Sande E., Novak S., Collins C., Welch C. B., Lusis A. J., et al. . 1998. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA. 95: 13018–13023. - PMC - PubMed
-
- Cases S., Stone S. J., Zhou P., Yen E., and Tow B.. 2001. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 276: 38870–38876. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
