

Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry
- PMID: 28374312
- PMCID: PMC5452613
- DOI: 10.1007/s13361-017-1635-x
Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry
Abstract
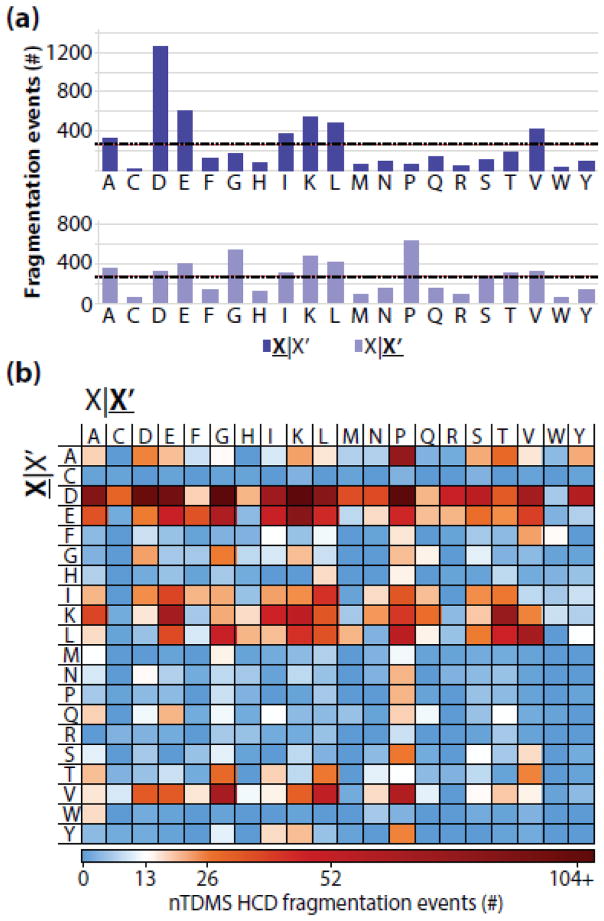
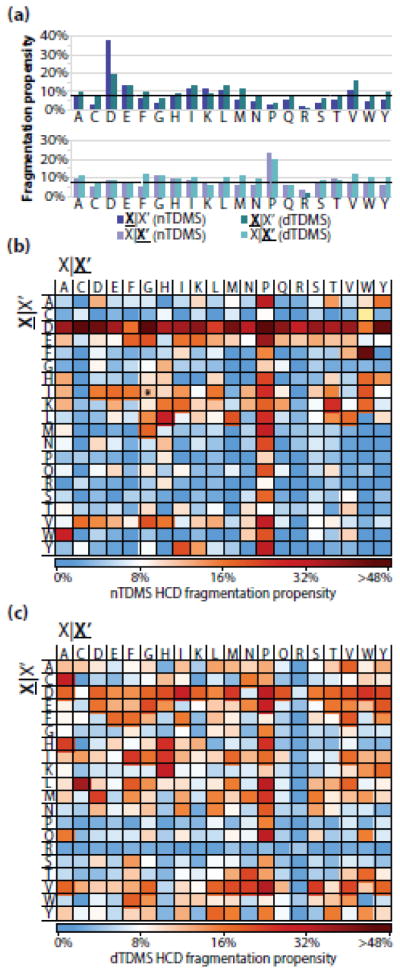
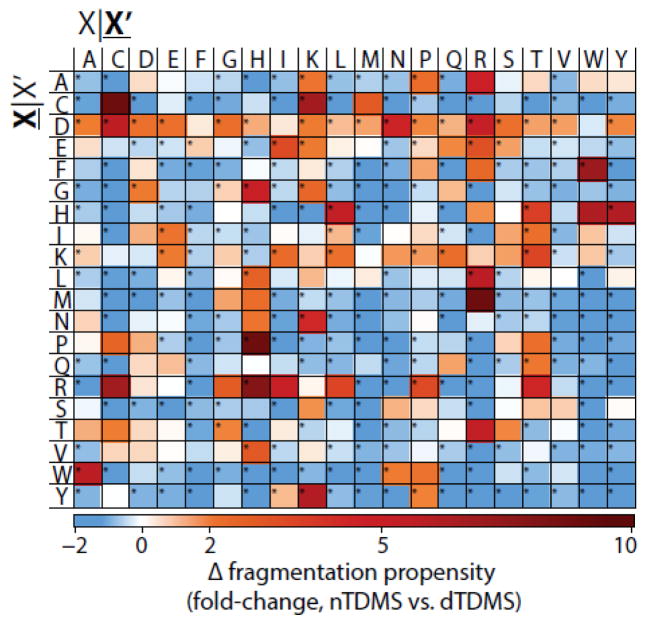
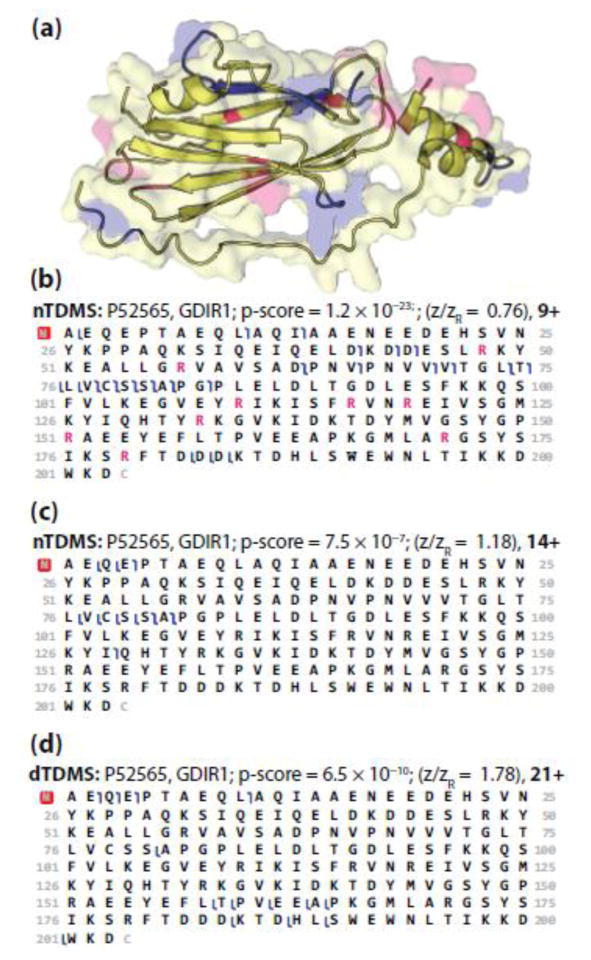
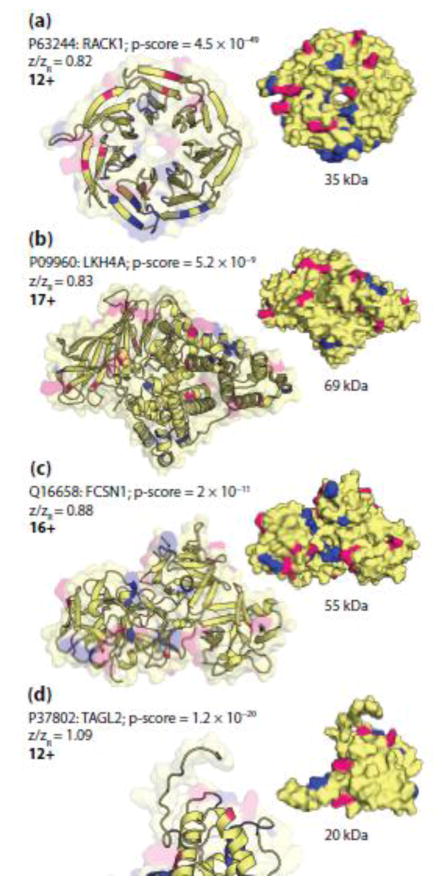
Fragmentation of intact proteins in the gas phase is influenced by amino acid composition, the mass and charge of precursor ions, higher order structure, and the dissociation technique used. The likelihood of fragmentation occurring between a pair of residues is referred to as the fragmentation propensity and is calculated by dividing the total number of assigned fragmentation events by the total number of possible fragmentation events for each residue pair. Here, we describe general fragmentation propensities when performing top-down mass spectrometry (TDMS) using denaturing or native electrospray ionization. A total of 5311 matched fragmentation sites were collected for 131 proteoforms that were analyzed over 165 experiments using native top-down mass spectrometry (nTDMS). These data were used to determine the fragmentation propensities for 399 residue pairs. In comparison to denatured top-down mass spectrometry (dTDMS), the fragmentation pathways occurring either N-terminal to proline or C-terminal to aspartic acid were even more enhanced in nTDMS compared with other residues. More generally, 257/399 (64%) of the fragmentation propensities were significantly altered (P ≤ 0.05) when using nTDMS compared with dTDMS, and of these, 123 were altered by 2-fold or greater. The most notable enhancements of fragmentation propensities for TDMS in native versus denatured mode occurred (1) C-terminal to aspartic acid, (2) between phenylalanine and tryptophan (F|W), and (3) between tryptophan and alanine (W|A). The fragmentation propensities presented here will be of high value in the development of tailored scoring systems used in nTDMS of both intact proteins and protein complexes. Graphical Abstract ᅟ.
Keywords: Fragmentation propensity; Native ESI; Native electrospray ionization; Native mass spectrometry; Residue fragmentation propensity; Tandem mass spectrometry; Top-down mass spectrometry.
Figures

References
-
- Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–258. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
