

Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer
- PMID: 28374746
- PMCID: PMC5518983
- DOI: 10.1038/cr.2017.51
Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer
Abstract
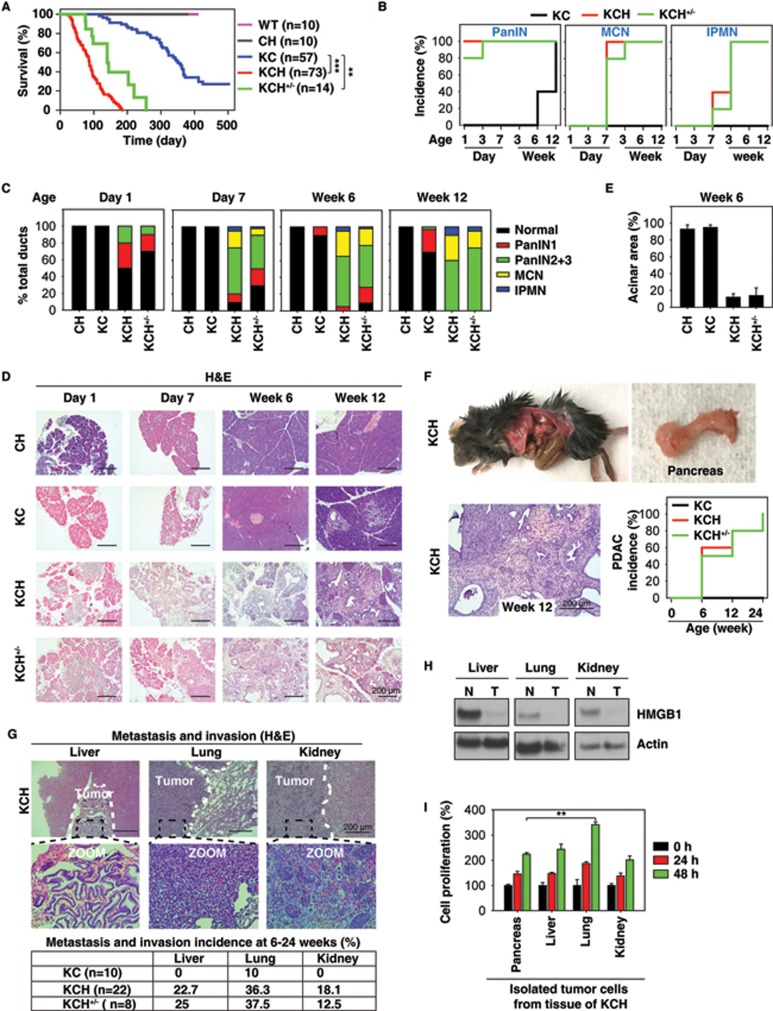
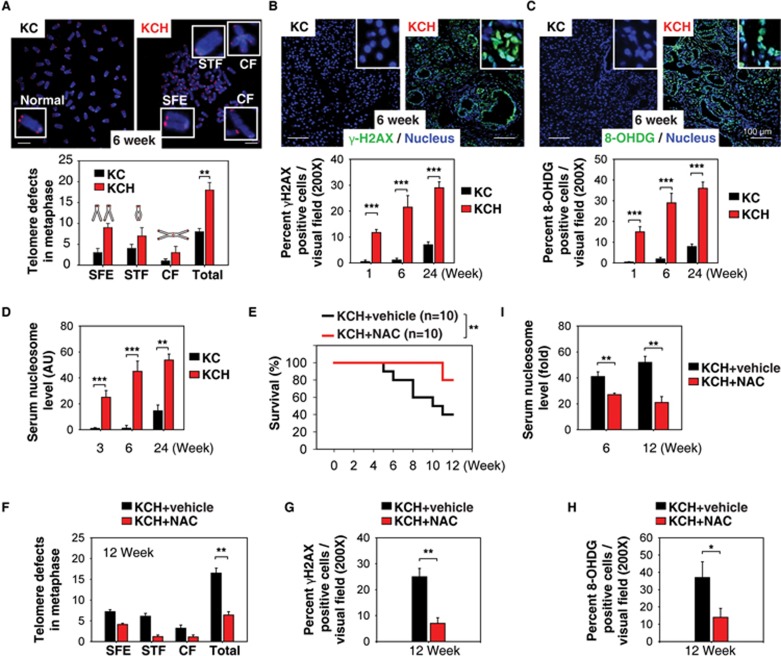
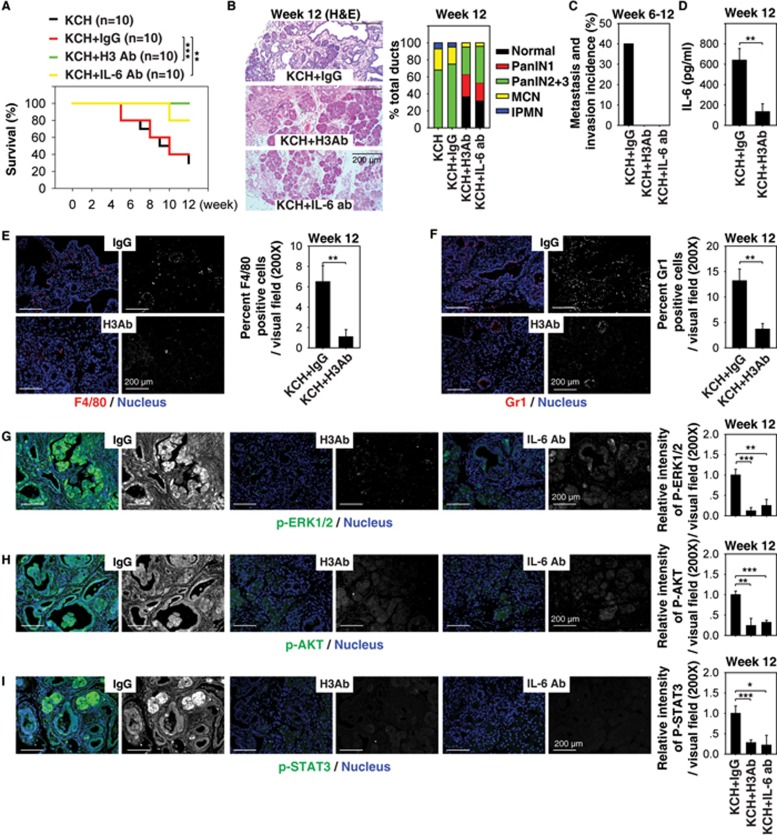
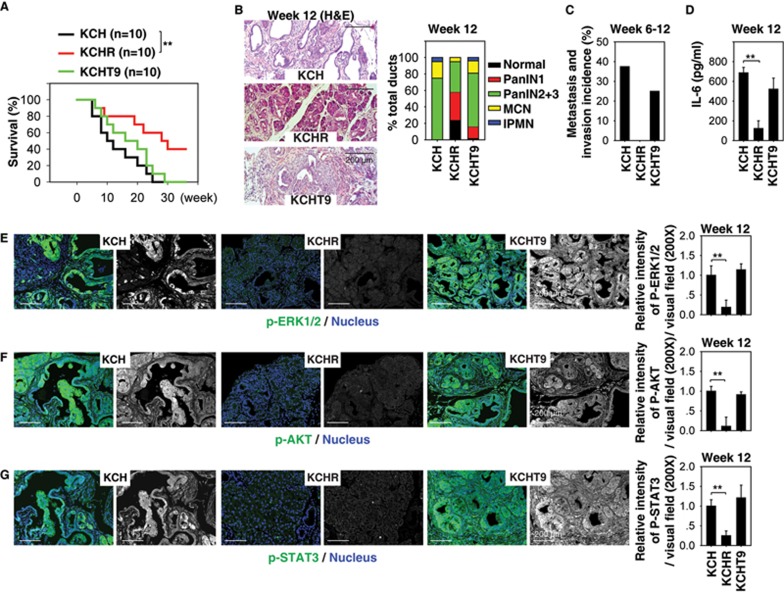
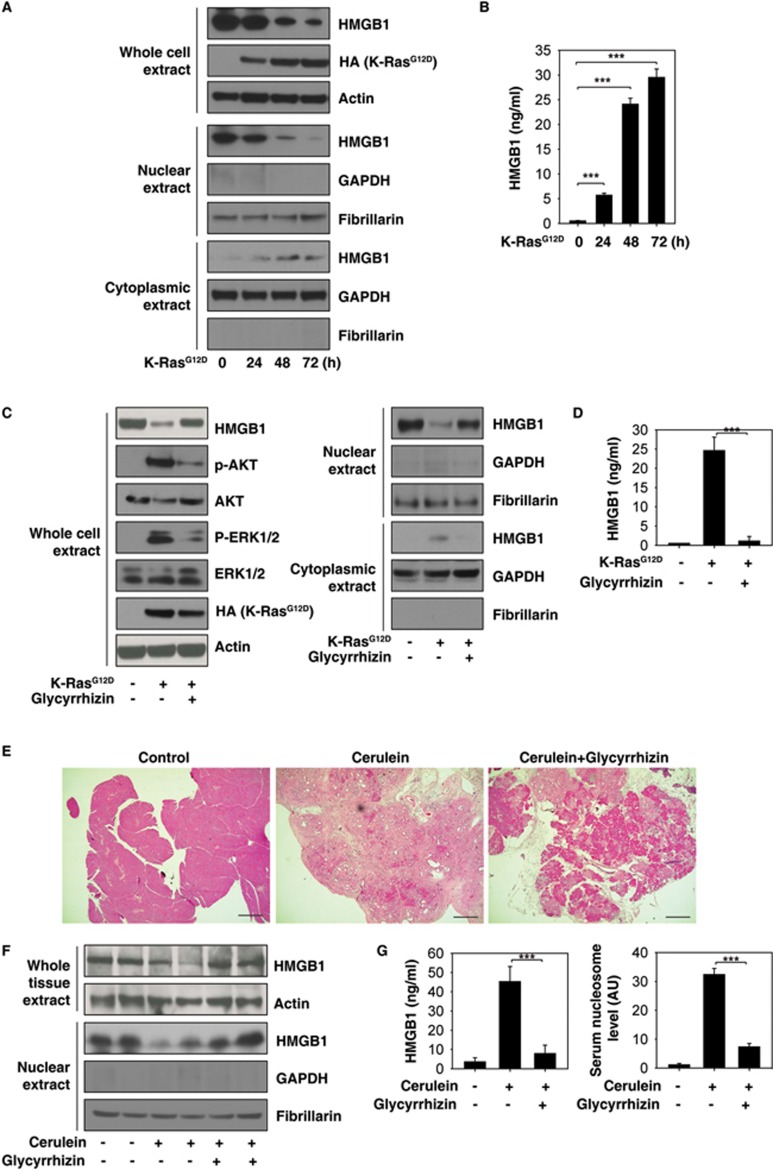
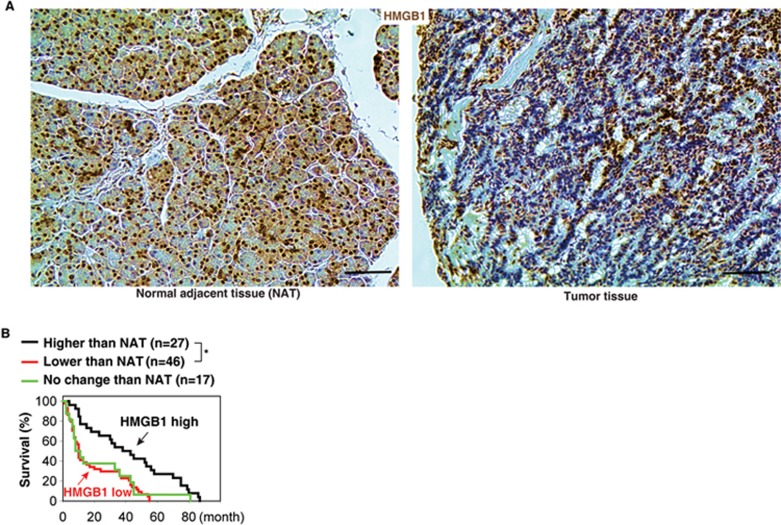
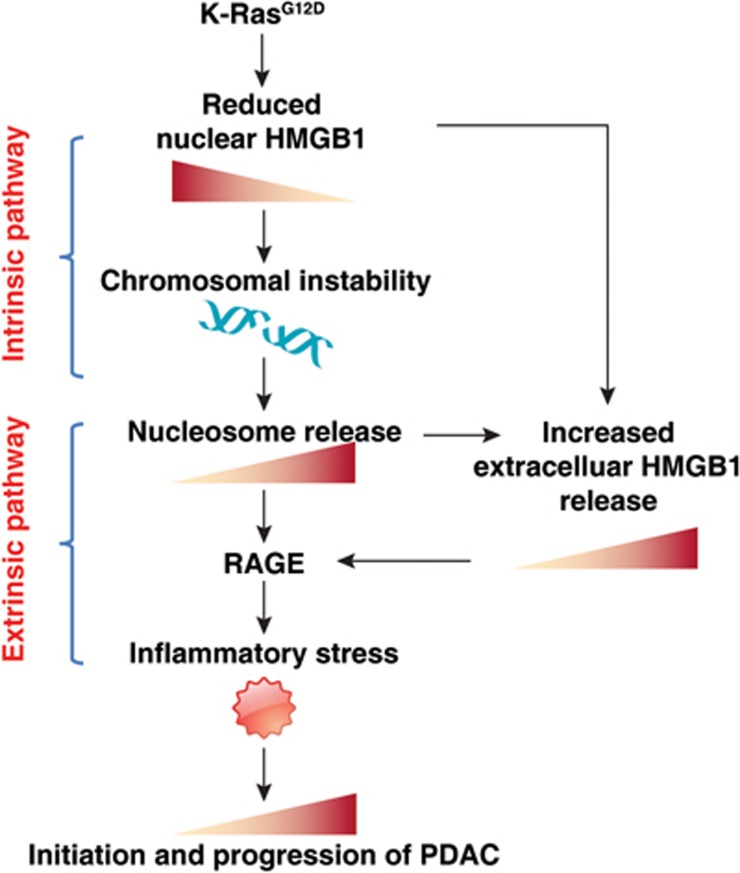
Pancreatic ductal adenocarcinoma (PDAC) driven by oncogenic K-Ras remains among the most lethal human cancers despite recent advances in modern medicine. The pathogenesis of PDAC is partly attributable to intrinsic chromosome instability and extrinsic inflammation activation. However, the molecular link between these two events in pancreatic tumorigenesis has not yet been fully established. Here, we show that intracellular high mobility group box 1 (HMGB1) remarkably suppresses oncogenic K-Ras-driven pancreatic tumorigenesis by inhibiting chromosome instability-mediated pro-inflammatory nucleosome release. Conditional genetic ablation of either single or both alleles of HMGB1 in the pancreas renders mice extremely sensitive to oncogenic K-Ras-driven initiation of precursor lesions at birth, including pancreatic intraepithelial neoplasms, intraductal papillary mucinous neoplasms, and mucinous cystic neoplasms. Loss of HMGB1 in the pancreas is associated with oxidative DNA damage and chromosomal instability characterized by chromosome rearrangements and telomere abnormalities. These lead to inflammatory nucleosome release and propagate K-Ras-driven pancreatic tumorigenesis. Extracellular nucleosomes promote interleukin 6 (IL-6) secretion by infiltrating macrophages/neutrophils and enhance oncogenic K-Ras signaling activation in pancreatic lesions. Neutralizing antibodies to IL-6 or histone H3 or knockout of the receptor for advanced glycation end products all limit K-Ras signaling activation, prevent cancer development and metastasis/invasion, and prolong animal survival in Pdx1-Cre;K-RasG12D/+;Hmgb1-/- mice. Pharmacological inhibition of HMGB1 loss by glycyrrhizin limits oncogenic K-Ras-driven tumorigenesis in mice under inflammatory conditions. Diminished nuclear and total cellular expression of HMGB1 in PDAC patients correlates with poor overall survival, supporting intracellular HMGB1 as a novel tumor suppressor with prognostic and therapeutic relevance in PDAC.
Figures
References
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5–29. - PubMed
-
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371:1039–1049. - PubMed
-
- Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol 2015; 12:319–334. - PubMed
-
- Cook N, Jodrell DI, Tuveson DA. Predictive in vivo animal models and translation to clinical trials. Drug Discov Today 2012; 17:253–260. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
