

Rad51 Degradation: Role in Oncolytic Virus-Poly(ADP-Ribose) Polymerase Inhibitor Combination Therapy in Glioblastoma
- PMID: 28376211
- PMCID: PMC6059185
- DOI: 10.1093/jnci/djw229
Rad51 Degradation: Role in Oncolytic Virus-Poly(ADP-Ribose) Polymerase Inhibitor Combination Therapy in Glioblastoma
Abstract
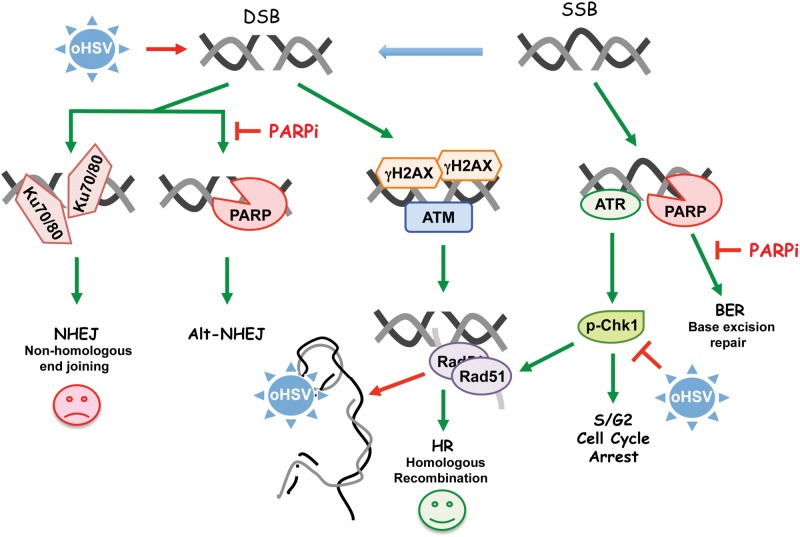
Background: Clinical success of poly(ADP-ribose) polymerase inhibitors (PARP i ) has been limited to repair-deficient cancers and by resistance. Oncolytic herpes simplex viruses (oHSVs) selectively kill cancer cells, irrespective of mutation, and manipulate DNA damage responses (DDR). Here, we explore potential synthetic lethal-like interactions between oHSV and PARP i .
Methods: The efficacy of combining PARP i , oHSV MG18L, and G47Δ in killing patient-derived glioblastoma stem cells (GSCs) was assessed using cell viability assays and Chou-Talalay synergy analysis. Effects on DDR pathways, apoptosis, and cell cycle after manipulation with pharmacological inhibitors and lentivirus-mediated knockdown or overexpression were examined by immunoblotting and FACS. In vivo efficacy was evaluated in two GSC-derived orthotopic xenograft models (n = 7-8 per group). All statistical tests were two-sided.
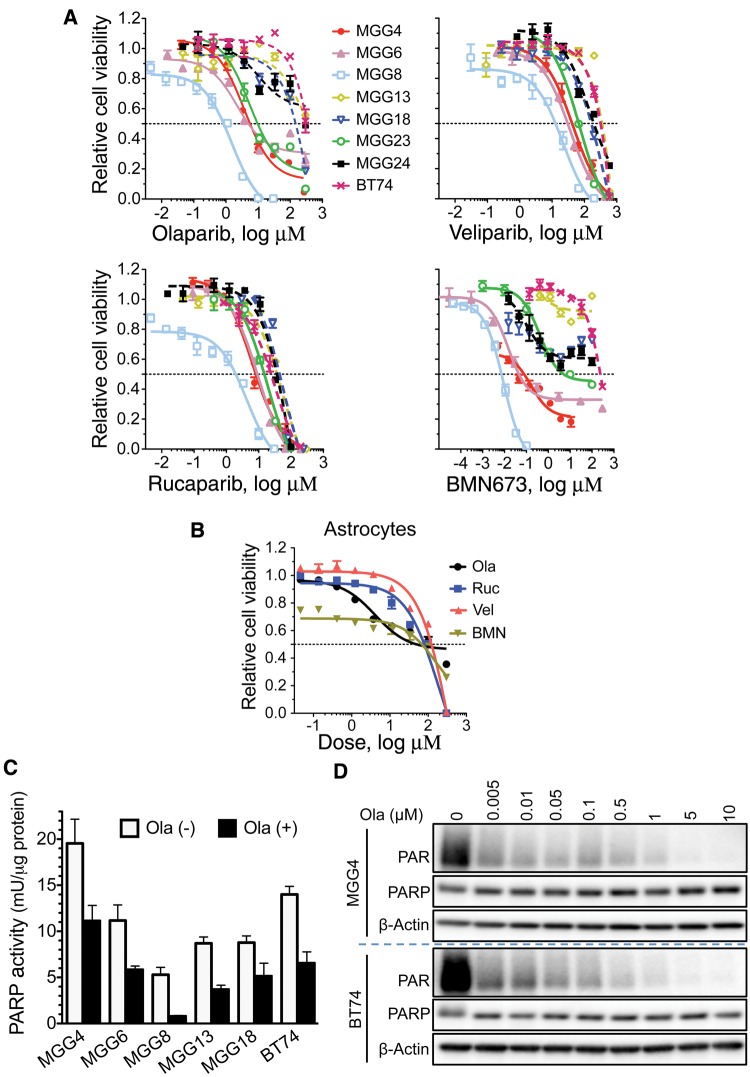
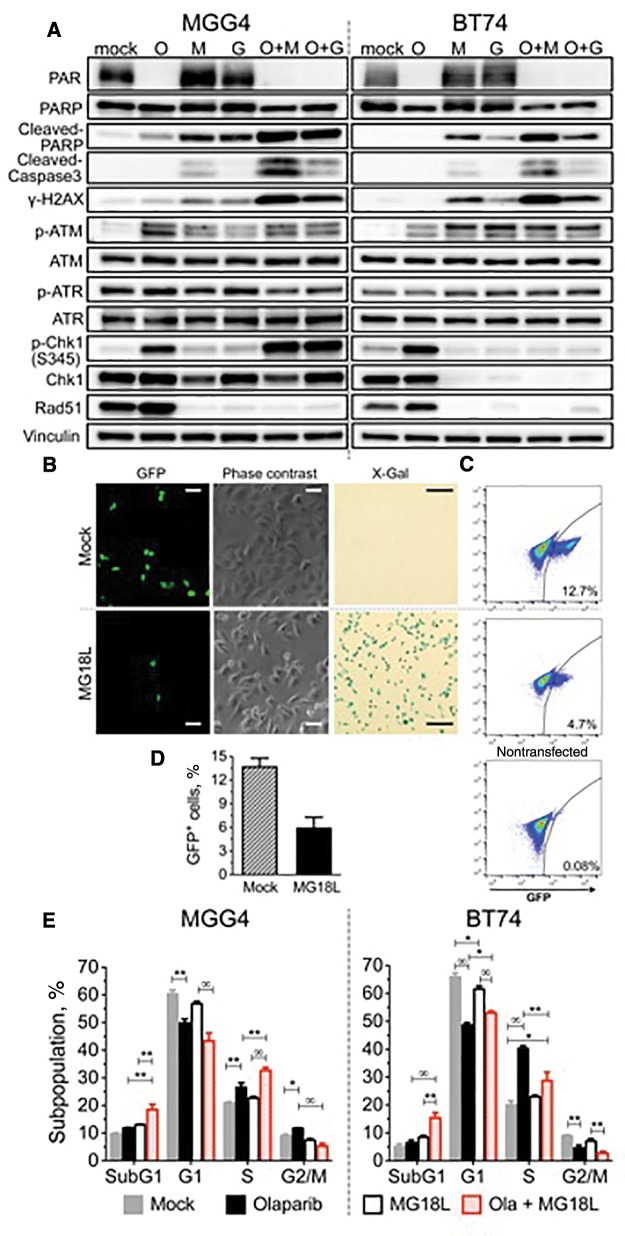
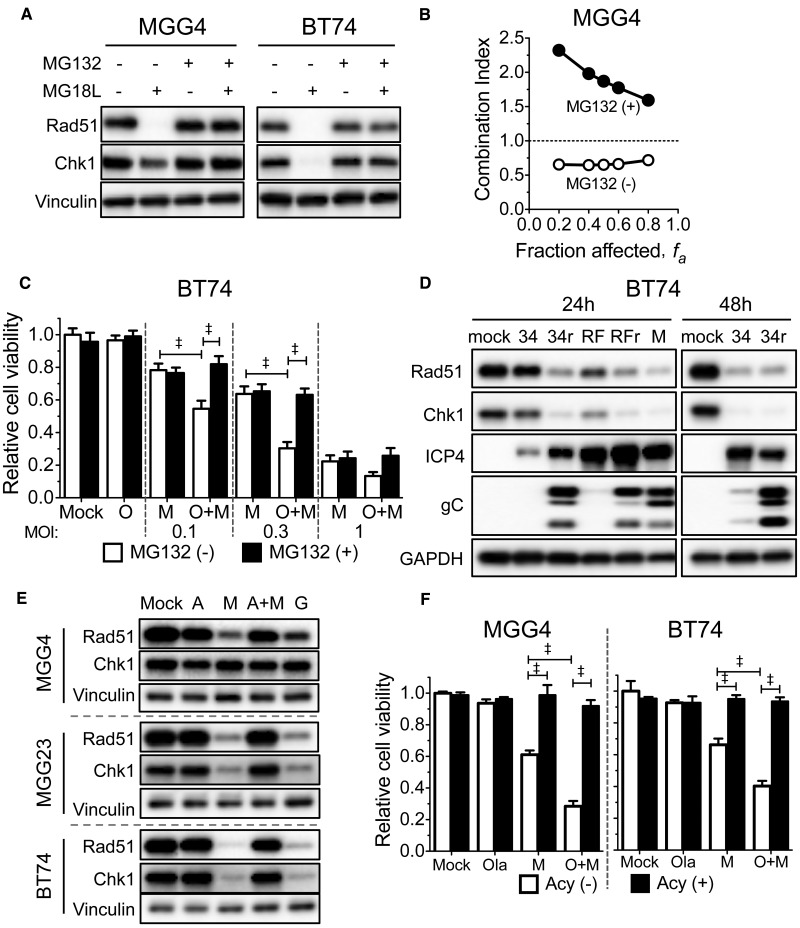
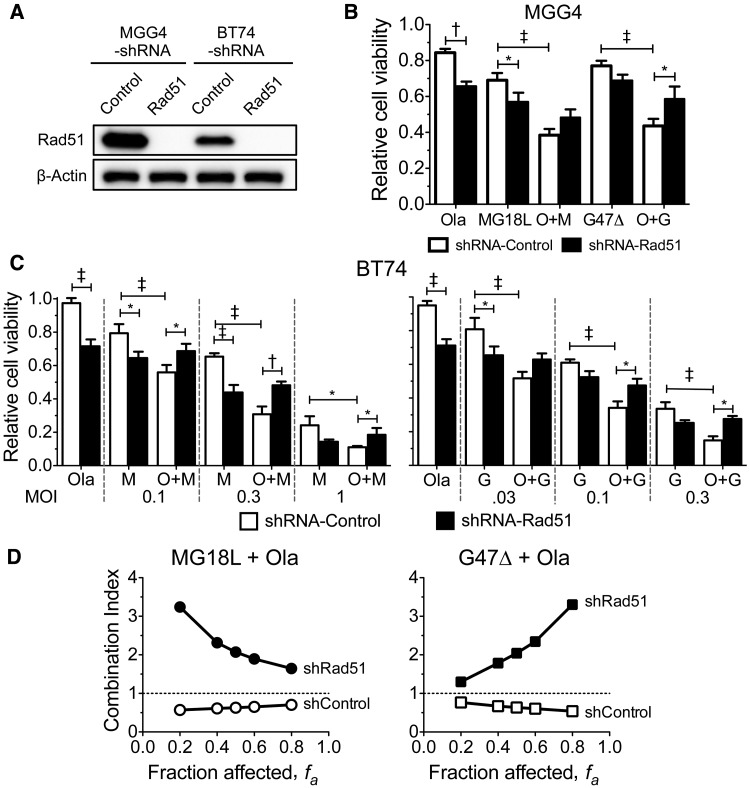
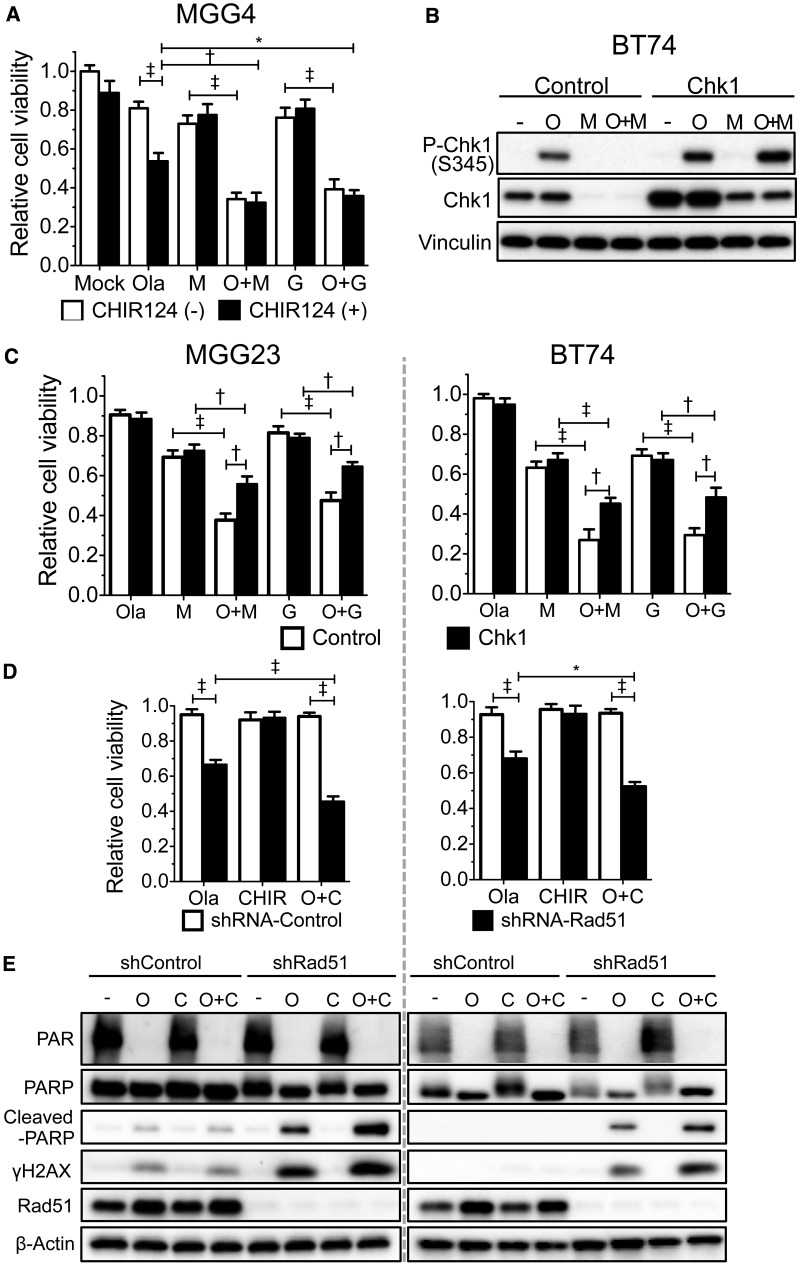
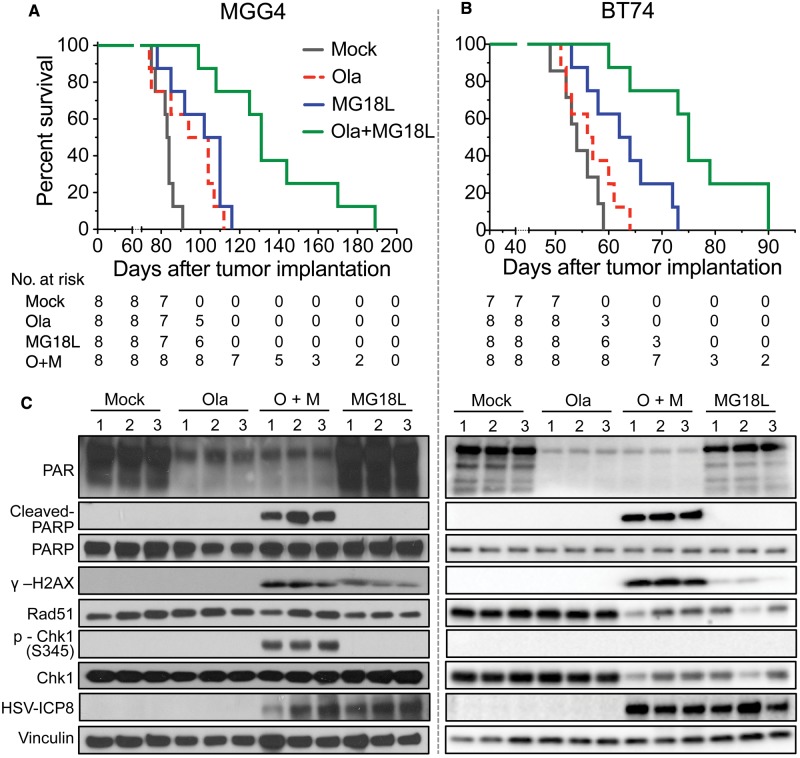
Results: GSCs are differentially sensitive to PARP i despite uniform inhibition of PARP activity. oHSV sensitized GSCs to PARP i , irrespective of their PARP i sensitivity through selective proteasomal degradation of key DDR proteins; Rad51, mediating the combination effects; and Chk1. Rad51 degradation required HSV DNA replication. This synthetic lethal-like interaction increased DNA damage, apoptosis, and cell death in vitro and in vivo. Combined treatment of mice bearing PARP i -sensitive or -resistant GSC-derived brain tumors greatly extended median survival compared to either agent alone (vs olaparib: P ≤.001; vs MG18L: P = .005; median survival for sensitive of 83 [95% CI = 77 to 86], 94 [95% CI = 75 to 107], 102 [95% CI = 85 to 110], and 131 [95% CI = 108 to 170] days and for resistant of 54 [95% CI = 52 to 58], 56 [95% CI = 52 to 61], 62 [95% CI = 56 to 72], and 75 [95% CI = 64 to 90] days for mock, PARPi, oHSV, and combination, respectively).
Conclusions: The unique oHSV property to target multiple components of DDR generates cancer selective sensitivity to PARP i . This combination of oHSV with PARP i is a new anticancer strategy that overcomes the clinical barriers of PARP i resistance and DNA repair proficiency and is applicable not only to glioblastoma, an invariably lethal tumor, but also to other tumor types.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures

Similar articles
-
Oncolytic herpes simplex viruses for the treatment of glioma and targeting glioblastoma stem-like cells.Front Cell Infect Microbiol. 2023 May 31;13:1206111. doi: 10.3389/fcimb.2023.1206111. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 37325516 Free PMC article. Review.
-
Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells.J Natl Cancer Inst. 2012 Jan 4;104(1):42-55. doi: 10.1093/jnci/djr509. Epub 2011 Dec 15. J Natl Cancer Inst. 2012. PMID: 22173583 Free PMC article.
-
A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells.Clin Cancer Res. 2011 Jun 1;17(11):3686-96. doi: 10.1158/1078-0432.CCR-10-3142. Epub 2011 Apr 19. Clin Cancer Res. 2011. PMID: 21505062 Free PMC article.
-
Restriction of Replication of Oncolytic Herpes Simplex Virus with a Deletion of γ34.5 in Glioblastoma Stem-Like Cells.J Virol. 2018 Jul 17;92(15):e00246-18. doi: 10.1128/JVI.00246-18. Print 2018 Aug 1. J Virol. 2018. PMID: 29793956 Free PMC article.
-
[Abnormalities of DNA repair and gynecological cancers].Bull Cancer. 2017 Nov;104(11):971-980. doi: 10.1016/j.bulcan.2017.09.007. Epub 2017 Oct 18. Bull Cancer. 2017. PMID: 29054544 Review. French.
Cited by
-
Oncolytic vesicular stomatitis virus alone or in combination with JAK inhibitors is effective against ovarian cancer.Mol Ther Oncol. 2024 Jun 8;32(3):200826. doi: 10.1016/j.omton.2024.200826. eCollection 2024 Sep 19. Mol Ther Oncol. 2024. PMID: 39006945 Free PMC article.
-
Budding yeast Rad51: a paradigm for how phosphorylation and intrinsic structural disorder regulate homologous recombination and protein homeostasis.Curr Genet. 2021 Jun;67(3):389-396. doi: 10.1007/s00294-020-01151-2. Epub 2021 Jan 12. Curr Genet. 2021. PMID: 33433732 Free PMC article. Review.
-
Histone deacetylase inhibitors enhance oncolytic herpes simplex virus therapy for malignant meningioma.Biomed Pharmacother. 2022 Nov;155:113843. doi: 10.1016/j.biopha.2022.113843. Epub 2022 Oct 8. Biomed Pharmacother. 2022. PMID: 36271587 Free PMC article.
-
Role of PARP Inhibitors in Glioblastoma and Perceiving Challenges as Well as Strategies for Successful Clinical Development.Front Pharmacol. 2022 Jul 6;13:939570. doi: 10.3389/fphar.2022.939570. eCollection 2022. Front Pharmacol. 2022. PMID: 35873570 Free PMC article. Review.
-
Oncolytic herpes simplex viruses for the treatment of glioma and targeting glioblastoma stem-like cells.Front Cell Infect Microbiol. 2023 May 31;13:1206111. doi: 10.3389/fcimb.2023.1206111. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 37325516 Free PMC article. Review.
References
-
- O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–560. - PubMed
-
- Dietlein F, Thelen L, Reinhardt HC.. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014;30(8):326–339. - PubMed
-
- Lord CJ, Tutt AN, Ashworth A.. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
