

Image analysis driven single-cell analytics for systems microbiology
- PMID: 28376782
- PMCID: PMC5379763
- DOI: 10.1186/s12918-017-0399-z
Image analysis driven single-cell analytics for systems microbiology
Abstract
Background: Time-lapse microscopy is an essential tool for capturing and correlating bacterial morphology and gene expression dynamics at single-cell resolution. However state-of-the-art computational methods are limited in terms of the complexity of cell movies that they can analyze and lack of automation. The proposed Bacterial image analysis driven Single Cell Analytics (BaSCA) computational pipeline addresses these limitations thus enabling high throughput systems microbiology.
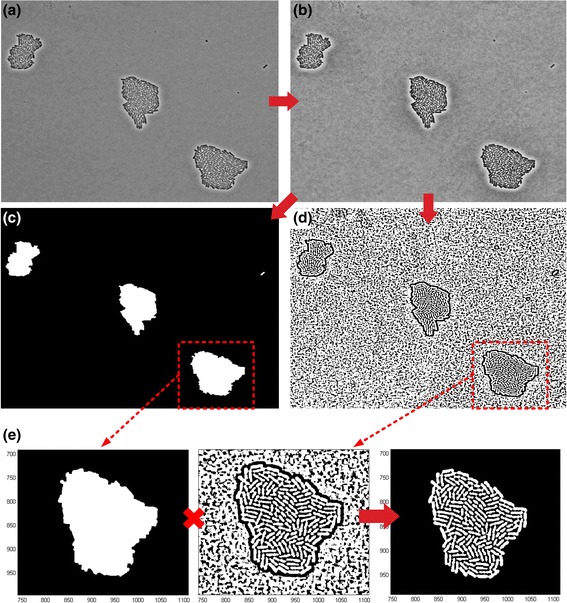
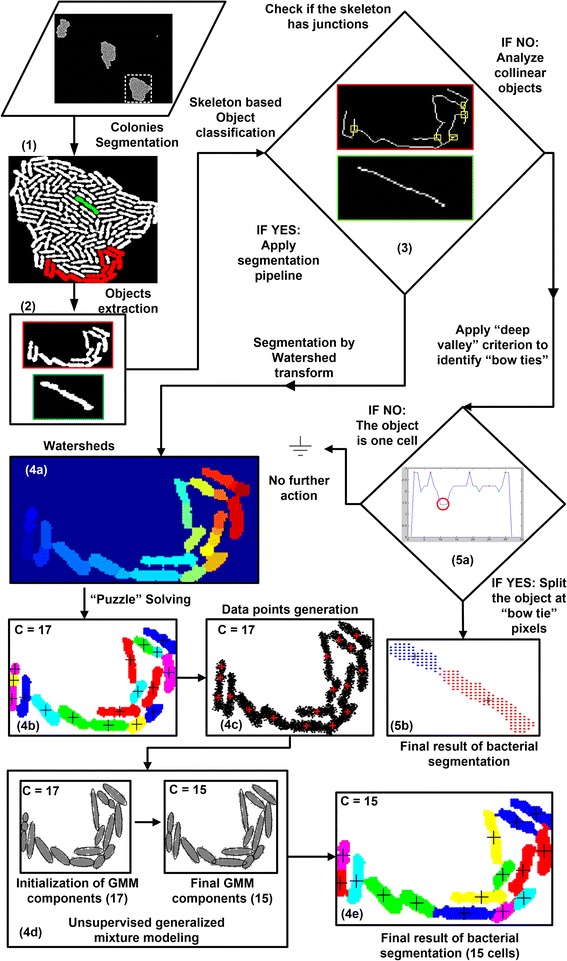
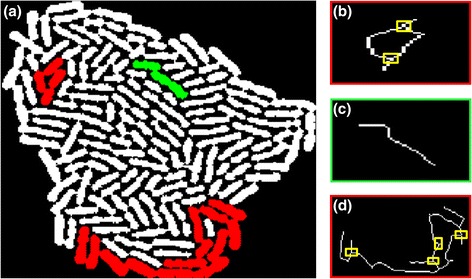
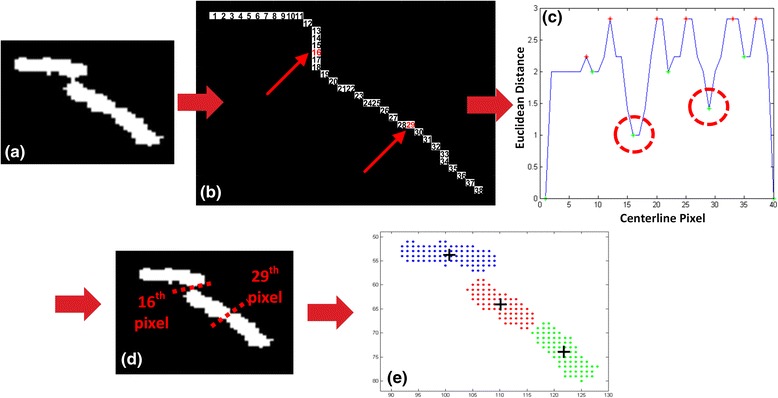
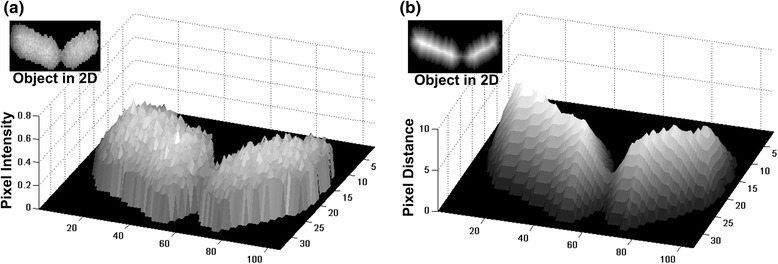
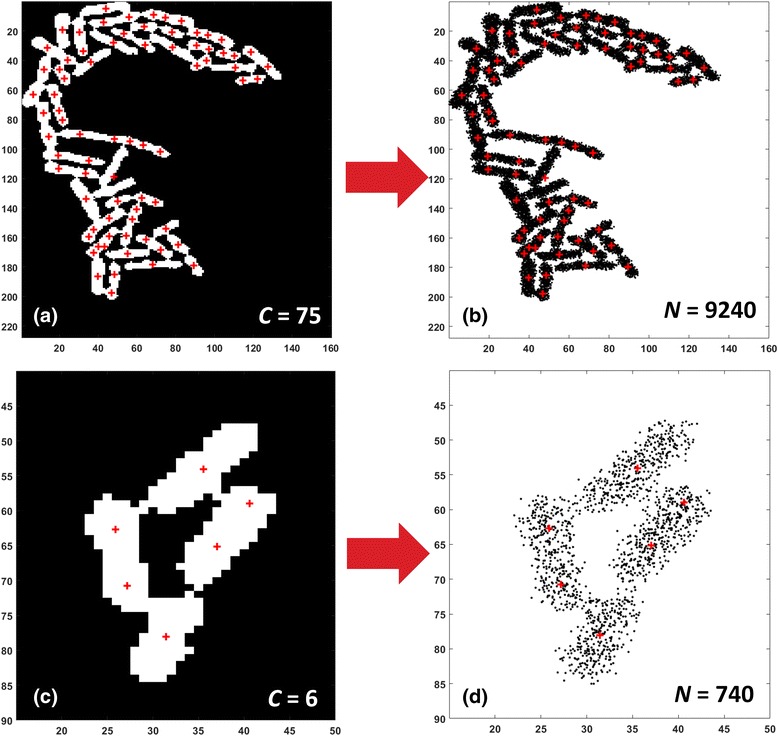
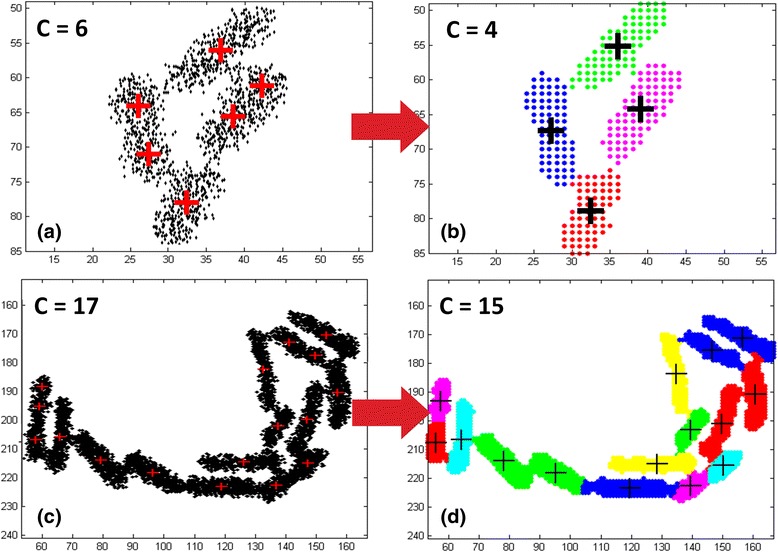
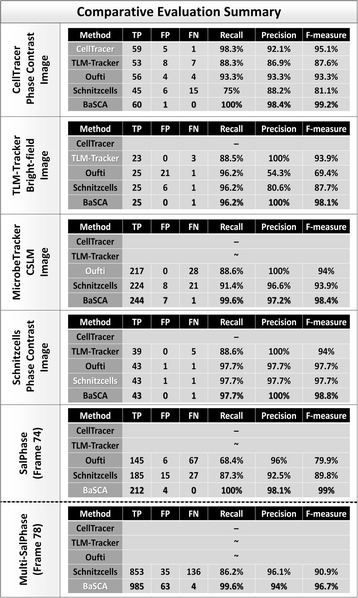
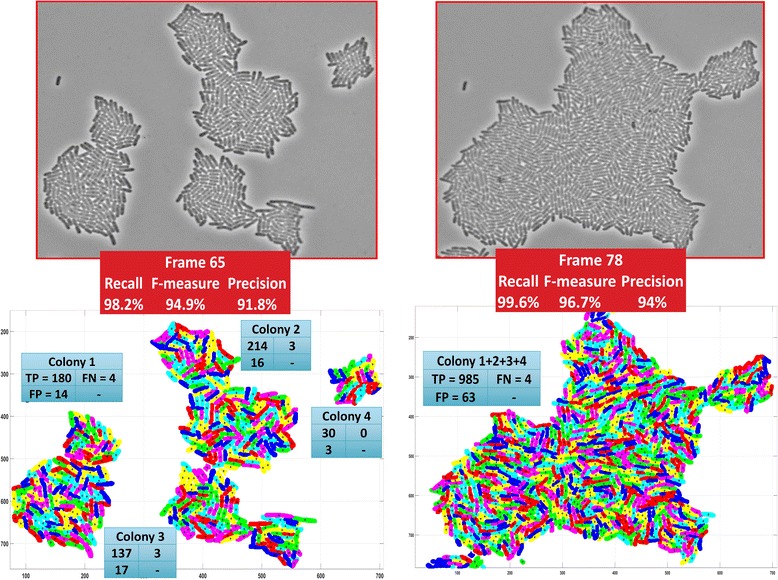
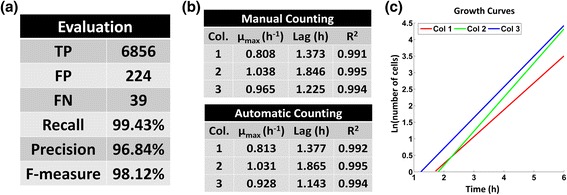
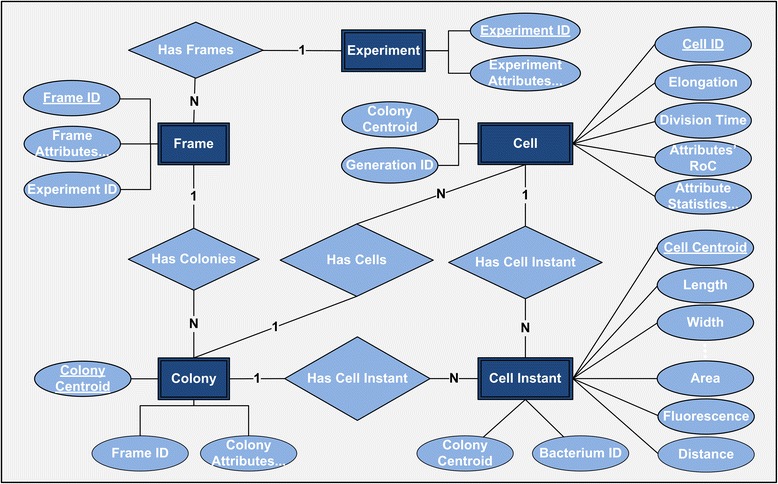
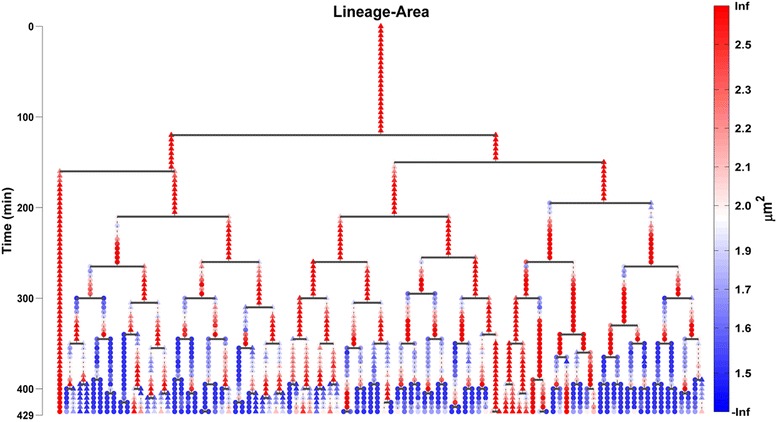
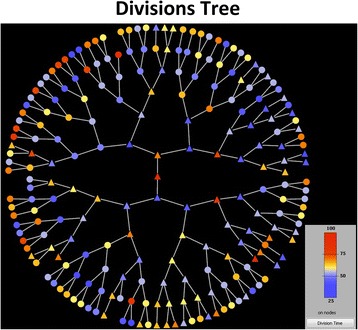
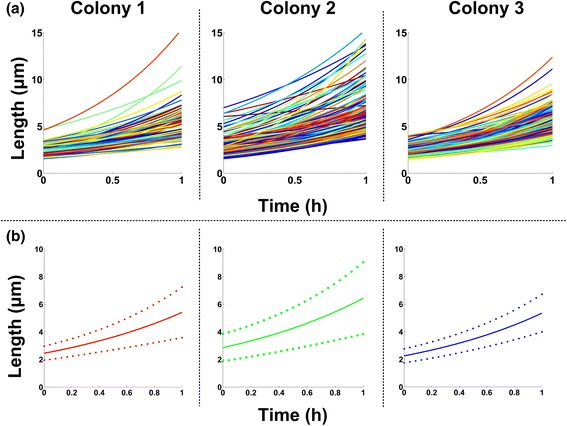
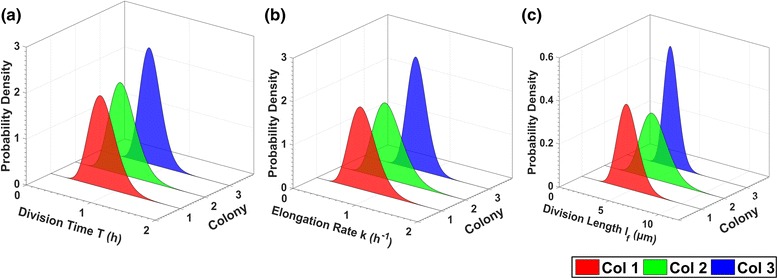
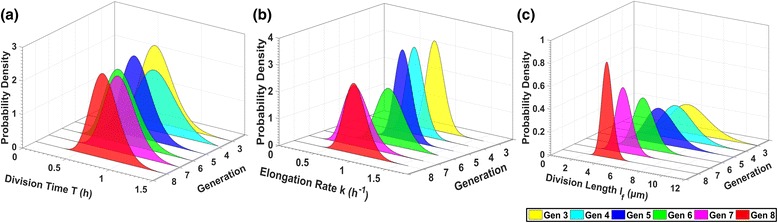
Results: BaSCA can segment and track multiple bacterial colonies and single-cells, as they grow and divide over time (cell segmentation and lineage tree construction) to give rise to dense communities with thousands of interacting cells in the field of view. It combines advanced image processing and machine learning methods to deliver very accurate bacterial cell segmentation and tracking (F-measure over 95%) even when processing images of imperfect quality with several overcrowded colonies in the field of view. In addition, BaSCA extracts on the fly a plethora of single-cell properties, which get organized into a database summarizing the analysis of the cell movie. We present alternative ways to analyze and visually explore the spatiotemporal evolution of single-cell properties in order to understand trends and epigenetic effects across cell generations. The robustness of BaSCA is demonstrated across different imaging modalities and microscopy types.
Conclusions: BaSCA can be used to analyze accurately and efficiently cell movies both at a high resolution (single-cell level) and at a large scale (communities with many dense colonies) as needed to shed light on e.g. how bacterial community effects and epigenetic information transfer play a role on important phenomena for human health, such as biofilm formation, persisters' emergence etc. Moreover, it enables studying the role of single-cell stochasticity without losing sight of community effects that may drive it.
Keywords: Bacterial image analysis; Cell segmentation; Colonies segmentation; Lineage tree construction; Machine learning; Single-cell analytics; Single-cell informatics; Time-lapse microscopy; Visualization.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
