

Gene Therapy for β-Hemoglobinopathies
- PMID: 28377044
- PMCID: PMC5417842
- DOI: 10.1016/j.ymthe.2017.03.024
Gene Therapy for β-Hemoglobinopathies
Abstract
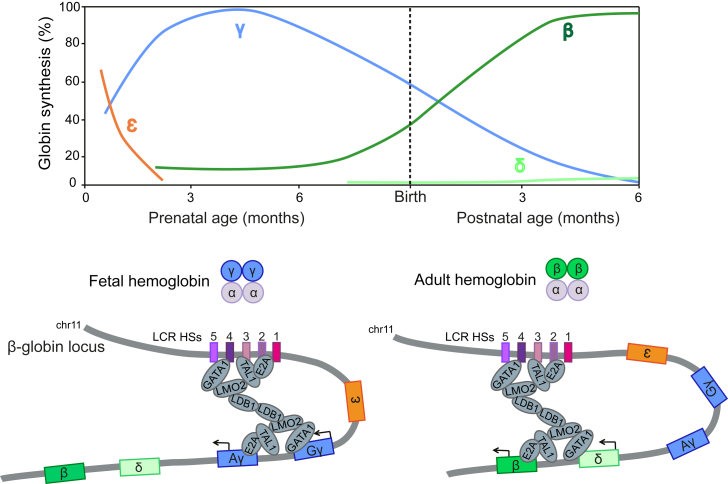
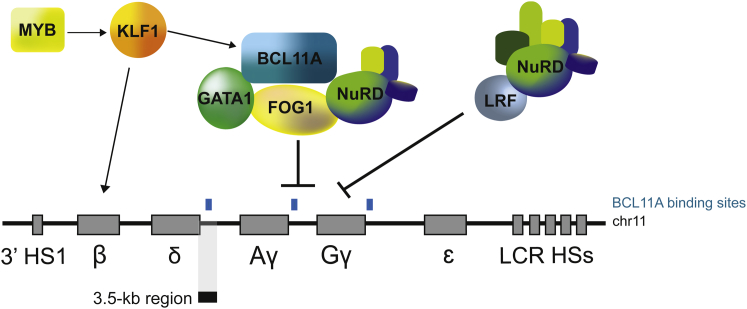
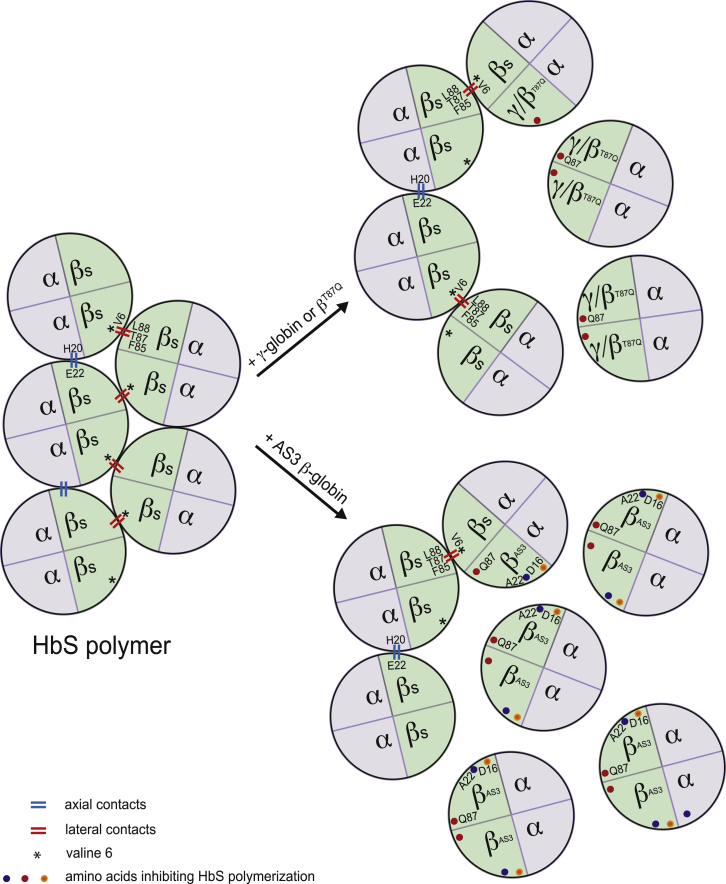
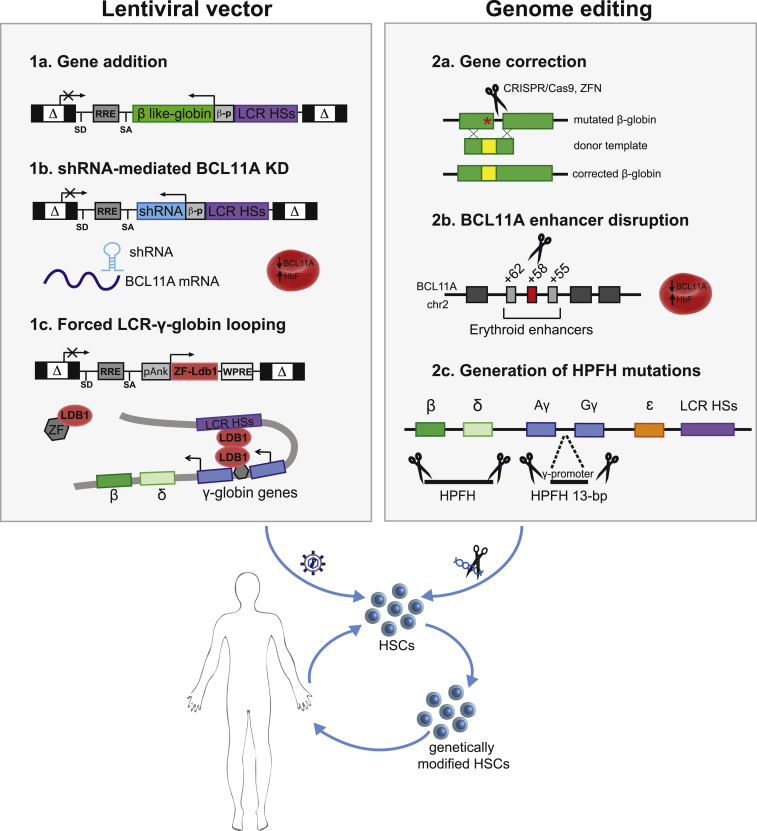
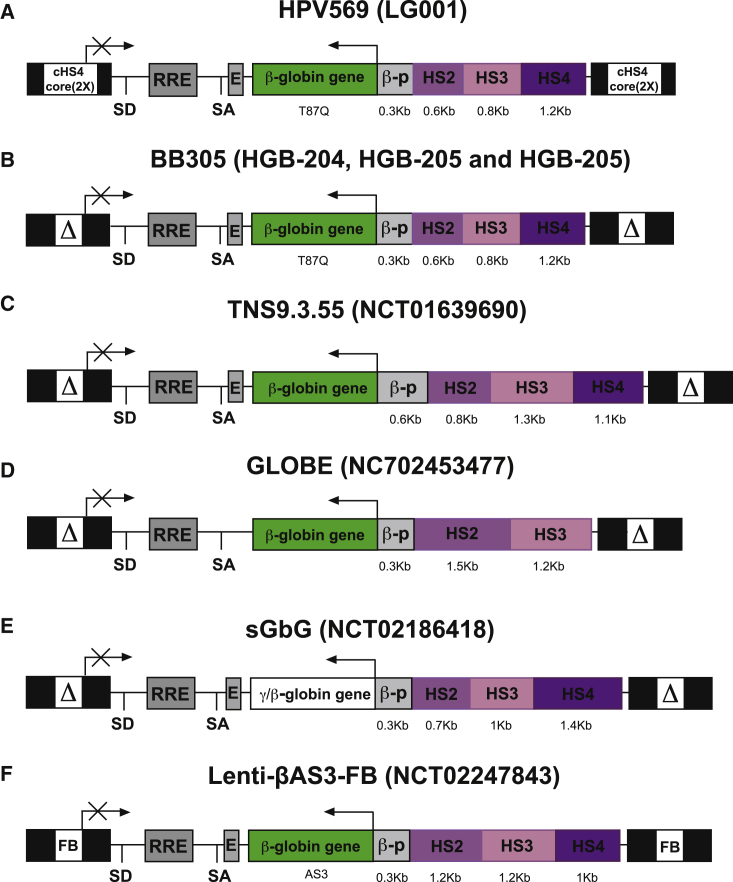
β-Thalassemia and sickle cell disease (SCD) are the world's two most widely disseminated hereditary hemoglobinopathies. β-Thalassemia originated in the Mediterranean, Middle Eastern, and Asian regions, and SCD originated in central Africa. However, subsequent population migration means that these two diseases are now global and thus constitute a growing health problem in many countries. Despite remarkable improvements in medical care for patients with β-hemoglobinopathies, there is still only one definitive treatment option: allogeneic hematopoietic stem cell (HSC) transplantation. The development of gene therapy for β-hemoglobinopathies has been justified by (1) the limited availability of human leukocyte antigen (HLA)-identical donors, (2) the narrow window of application of HSC transplantation to the youngest patients, and (3) recent advances in HSC-based gene therapy. The huge ongoing efforts in translational medicine and the high number of related publications show that gene therapy has the potential to become the treatment of choice for patients who lack either an HLA genoidentical sibling or an alternative, medically acceptable donor. In this dynamic scientific context, we first summarize the main steps toward clinical translation of this therapeutic approach and then discuss novel lentiviral- and genome editing-based treatment strategies for β-hemoglobinopathies.
Keywords: gene therapy; hematopoietic stem cell; hemoglobinopathies; sickle cell disease; thalassemias.
Copyright © 2017 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Zhou W., Zhao Q., Sutton R., Cumming H., Wang X., Cerruti L., Hall M., Wu R., Cunningham J.M., Jane S.M. The role of p22 NF-E4 in human globin gene switching. J. Biol. Chem. 2004;279:26227–26232. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
